Figure 16.1. EEG showing classic hypsarrhythmia consisting of high-amplitude polymorphic delta waves and multiregional spike waves.
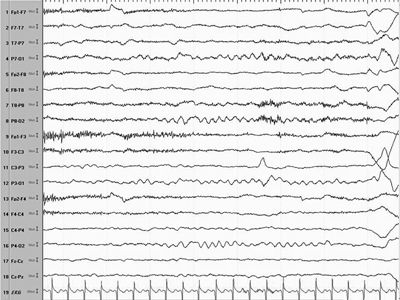
Figure 16.2. EEG of a patient in Figure 16.1 shows normal background and resolution of hypsarrhythmia within 4 weeks on ACTH.
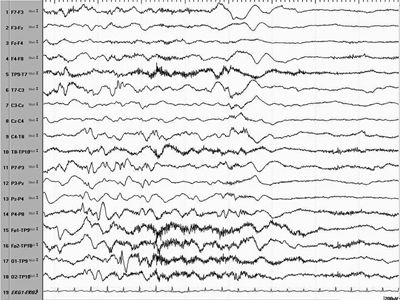
Figure 16.3. Electrodecremental pattern associated with an IS preceded by a broad central slow wave.
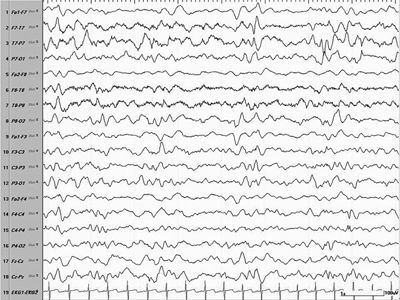
Figure 16.4. EEG showing a modified hypsarrhythmia pattern consisting of more lateralized high-amplitude multifocal spikes over the left hemisphere, suggesting a structural lesion.
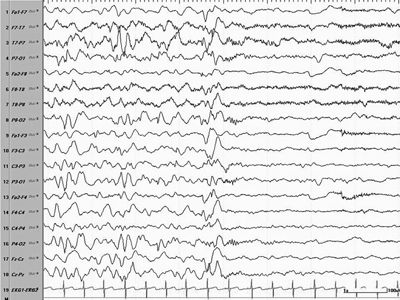
Figure 16.5. EEG showing an ictal change associated with an ES in the form of a generalized wave followed by an electrodecremental pattern.
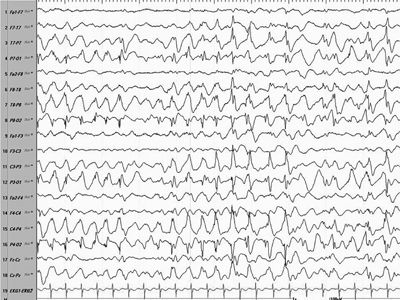
Figure 16.6. Modified hypsarrhythmia in an older child with more synchronized posterior hypersynchronous activity.
Rarely, the interictal EEG may be normal early in the onset of ES and should, therefore, be repeated serially at close intervals if there is a high index of suspicion for the diagnosis of ES (17). By the same token, ES caused by a localized cortical pathology involving a single lobe or hemisphere may not be associated with hypsarrhythmia, and focal slow-wave activity or a persistently interictal spike focus localizing to one brain region even in the presence of multifocal spikes may point to focal epileptiform pathology such as cortical dysplasia, porencephaly, or a developmental tumor. Hypsarrhythmia may precede the presentation of ES and be found incidentally with routine EEG surveillance or patterns that evolve into hypsarrhythmia may be present prior to evolution of ES that may allow early detection of infants who are at risk for ES (72–74).
The background activity never approaches normal frequencies or amplitudes and is characteristically of high voltage (500 to 1000 mV), disorganized, and asynchronous with a waxing and waning quality (2).
The sleep–wake cycle has a significant effect on the manifestation of the pattern of hypsarrhythmia. During non–rapid eye movement (REM) sleep, there may be fragmentation of the hypsarrhythmic pattern, while by the end of REM sleep or during arousal from sleep, near-normal activity may occur; this “pseudonormalization” may also immediately precede a cluster of spasms (75,76). Long-term monitoring has disclosed variable patterns throughout the day, with more hypsarrhythmia noted in slow-wave sleep and less in REM sleep (22). Fast-wave bursts were seen during REM sleep in 35% of patients with spasms, sometimes occurring periodically until clinical spasms appeared and the patient awakens (21). Spasms may start subclinically in REM sleep (76).
Any variation in the characteristic hypsarrhythmic pattern as described above has been termed modified hypsarrhythmia and includes background synchronization, very focal features, voltage asymmetries, generalized background burst suppression, and slow waves without spikes (17). In an analysis of pretreatment electroencephalograms, the modified pattern occurred in up to 36 (69%) of 53 patients with spasms; cortical dysplasia was associated with hemihypsarrhythmia or burst suppression (77).
Regarding the role of the EEG as a prognostic tool, a burst–suppression pattern, as seen in children with Ohtahara syndrome, suggests a poor prognosis; a lower-voltage EEG may indicate a better outcome (77), as may preservation of hypsarrhythmia between spasms (28,54), faster background activity, and absence of postictal electrodecremental responses (as described in the following paragraph). However, children with more typical features of hypsarrhythmia such as disorganization, slowing, high-amplitude, spike, and electrodecremental discharges independent of seizures, absent normal sleep architecture, burst suppression, hemihypsarrhythmia, occipital hypsarrhythmia, interhemispheric asymmetry, and interhemispheric synchronization may predict a more severe developmental and epilepsy seizure prognosis (77). In late-onset ES, a more organized electroencephalographic background predicts better development while persistent hypsarrhythmia and a disorganized background may be a risk factor for poorer prognosis (78).
The most common ictal discharge that has been described consists of an initial multiphasic, high-amplitude slow wave, sometimes of positive polarity or less frequently a de novo low-amplitude, brief, fast frequency discharge (19,28). The generalized positive slow waves are followed by attenuation or an electrodecremental response, observed mainly at PZ (parietal midline), FZ/PZ, or FZ (frontal midline) with some variable degree of laterality (17,23). Because the decremental activity follows the slow wave and clinical spasm, it most likely is a postictal phenomenon (19,28), and the slow wave corresponds to the actual ES. An electrodecremental response can also be seen in the absence of a clinical seizure (12). Slow-sharp and slow-wave complexes, although less frequent with spasms, differ from the elongated appearance of those in myoclonic seizures (28). Diffuse attenuation, generalized spike wave, paroxysmal fast activity or fast frequencies, and slow wave are also associated with ES. There is no correlation between the semiology of the spasms and the different ictal patterns. The ictal pattern usually is brief, only lasting for approximately 1 second, while longer patterns are usually also associated with behavioral arrest (22). EMG shows that the axial muscles contract earlier than the limb muscles and the head earlier than the arms (23). A review of the EEG and behavioral changes before and between the spasms suggests that a cluster of spasms may represent a single sustained ictal event rather than brief, repetitive seizures (79). Pseudonormalization and high-amplitude slowing may precede the spasms and are associated with decreased activity and interaction (76), while subclinical discharges without clinical manifestations are also possible at the end of a spasm cluster; however, surface EMG may confirm subclinical contractions without actual perceptible movement on video-EEG during the subclinical discharges (28).
Fast activity is often associated with tonic spasms with sustained tonic muscle contraction and may be more common in asymmetric spasms, suggesting a cortical onset for spasms (76,78,80–82). Fast activity can also fade during repeated spasms or in partial seizures that occur with spasms (83).
CURRENT MODELS AND THEORIES OF THE PATHOPHYSIOLOGY OF ES
Several promising animal models to study the pathophysiology of ES have recently emerged. However, an ideal model that recapitulates every aspect of human ES does not yet exist. The ideal model would develop ES at the appropriate age and possess an EEG background similar to hypsarrhythmia; the extensor or flexor spasms would occur in clusters and have an associated slow wave followed by an electrodecrement, and the response to currently available medications would mimic that seen in humans. Some of the issues are interspecies differences in brain development and the lack of comparative developmental biomarkers across the species for variable seizure phenotypes and EEG signatures. However, each model will hopefully add another piece to the puzzle to give a clearer picture that will potentially translate experimental findings into clinically useful therapies. Several different models have been recently reviewed: (i) the corticotropin-releasing hormone (CRH) model examining the role of stress and response to adrenocorticotropic hormone (ACTH) in the developing brain; (ii) the N-methyl-d-aspartic acid model examining cryptogenic IS; (iii) the tetrodotoxin model examining hyperexcitability provoked by a decrease of neuronal activity; (iv) the multiple hit model examining cortical and subcortical lesions mimicking symptomatic IS; (v) the aristaless gene (ARX) mutation models, a genetic knockout model and a knockin model that recapitulates human mutations of ARX with an IS and MR phenotype and may prove the hypothesis that a deficiency of cortical interneuronal GABAergic inhibition (interneuonopathy) underlies developmental epileptic encephalopathies; and lastly (vi), the Down syndrome model, Ts65Dn Mouse model, that suggests that GABAB receptor alterations (agonists) may be a spasm-provoking mechanism (84,85).
From a recent human tissue study of four infants with FCD and IS treated with surgical resection, GABAA receptor abnormalities have been described using a novel technique of electrophysiologic recording after oocyte membrane injection from cortical brain tissue of these infants. Characterization of the cortical GABAA receptor properties demonstrated unaltered intrinsic physiology but altered neuromodulation to neurosteroids and zinc, which parallels the response of IS to ACTH (86).
Current theories on the generation of ES are that they represent a nonspecific age-dependent reaction of the immature brain to injury involving subcortical structures that acts diffusely on the cortex, leading to the hypsarrhythmic electroencephalogram pattern and the generalized spasms (12). Individual case reports have described abnormalities in the pons and involvement of the serotonergic, noradrenergic, or cholinergic neurons in the brainstem nuclei to support this assumption (87–89). Brainstem origin has also been postulated on the basis of abnormalities in brainstem auditory-evoked responses in patients with spasms and disruptions of REM sleep (90–92). In addition, ES and hypsarrhythmia have been noted in children with severe cortical injury or absence of cortex, supporting this theory. Because hypsarrhythmia occurs mainly during sleep and the brainstem controls sleep cycles, this sleep association again suggests that the brainstem plays a role in the manifestations of ES (92,93).
The frequent intermixture of focal seizures with generalized or asymmetric spasms suggests a cortical–subcortical interaction, a hypothesis supported by the effectiveness of cortical resection in controlling generalized ES (19). In other words, the cortical lesion interacts with developing brainstem pathways, causing motor spasms that are similar to startle or cortical reflex myoclonus (83,94).
Further supporting the brainstem hypothesis are results of PET scans in patients with ES showing hypermetabolism of the lenticular nuclei (95). That serotonergic ([11C]-methyl-l-tryptophan) and α-aminobutyric acid (GABA)-ergic ([11C]-flumazenil) tracers may be more effective than FDG-PET in defining a focus in patients with ES also suggests brainstem involvement, because the raphe–cortical or striatal projections use serotonin as a neurotransmitter, and these pathways cause the diffuse hypsarrhythmic patterns on EEG (41). On ictal single photon emission computed tomography (CT) studies, both subcortical and cortical structures were activated (96).
The response to corticotropin suggests involvement of the hypothalamus and pituitary–adrenal axis. Baram has proposed that a nonspecific stressor releases the proconvulsant CRH, which may be the final common pathway for the multitude of etiologies of ES (97). CRH causes severe seizures and death in neurons associated with learning and memory, and its effects are especially important in infants because CRH receptors are most abundant during the early developmental period (98). To support the hypothesis that corticotropin inhibits the release and production of CRH through a negative feedback mechanism, Nagamitsu et al. (99) measured CSF levels of β-endorphin (also derived from a common precursor of corticotropin), corticotropin, and CRH (which releases both corticotropin and β-endorphin) in 20 patients with spasms. The CSF levels of β-endorphin and corticotropin were lower than in controls, as was the CRH level, although not significantly. Riikonen (63) observed that CSF corticotropin levels were higher in infants with cryptogenic than symptomatic spasms.
Because ES typically begin at the time when the first immunizations are administered, the question has been whether the association is causative or coincidental. Numerous anecdotal reports have noted the appearance of ES within a few hours to a few days after a diphtheria, pertussis, tetanus vaccination, although all controlled studies to date have failed to demonstrate any association (100–102). Some proposed immunologic mechanisms have been based on antibodies to brain tissue in blood samples from patients with ES (103,104), or increased numbers of activated B and T cells in the blood (83), or increased levels of HLA-DRw52 antigen (34). Calcium-mediated mechanisms have been postulated, but further studies are needed to substantiate this (105). mTOR pathways have also been implicated in TSC patients as well as children with cortical dysplasia and hemimegencephaly (106).
DIFFERENTIAL DIAGNOSIS AND EVALUATION
The true epileptic nature of spasms may be easily missed in the beginning and considered to be colic, gastroesophageal reflux, or paroxysmal crying (16). Other paroxysmal disorders that may mimic ES include benign myoclonus of infancy in which the interictal and ictal EEGs are normal, including in sleep; hyperekplexia, in which the jerks may be triggered by touching the nose; Sandifer syndrome due to gastroesophageal reflux, paroxysmal tonic upward gaze, jactatio capitis, spasmus nutans, breath-holding spells, and infantile gratification behavior (16).
West syndrome is the classic epileptic encephalopathy of infancy associated with ES and is characterized by a triad of ES, hypsarrhythmia, and developmental failure or regression. Over the years, salaam seizures, jackknife seizures, axial spasms, periodic spasms, and serial spasms have been used as synonyms for ES.
The age of onset is typically between 4 and 8 months (17), but ES can occur as early as 2 weeks or as late as 18 months of age (12,107) and, rarely, can begin in adulthood. In some studies (107), late-onset spasms may be cryptogenic or associated with cortical dysplasia, hypoxic–ischemic encephalopathy, or genetic anomalies and were refractory to medications (17). Late-onset spasms may be intermixed with atonic, tonic, partial, myoclonic, or generalized tonic–clonic seizures or atypical absences. The characteristic spasms generally resolve spontaneously or evolve to other generalized seizure types or focal seizures but may persist as ES in 15% to 23% of patients beyond 3 to 7 years of age (35,36).
Watanabe has suggested that a subset of the cryptogenic group may be truly an “idiopathic” form of West syndrome (54). These patients have normal development, and the spasms usually remit after a short period. Developmental regression and focal interictal EEG abnormalities are usually not present; symmetric hypsarrhythmia reappears between each spasm in a cluster; and a family history of seizures is common. This group may represent from approximately half to 80% of the cryptogenic cases (25,97), referred to as “unknown” in the updated classification.
West syndrome may eventually evolve into LGS in many children ranging from 3% to 54% in various studies (108,109). Tonic seizures usually coexist with and are more marked during this stage of the syndrome. Seizure clustering is seen more frequently in West syndrome, but less frequently in LGS (23). Clusters of ES will usually become single spasms in LGS in parallel, with the changed interictal pattern from hypsarrhythmia to a generalized slow spike-and-wave pattern at 1 to 2.5 Hz (94).
The epileptic encephalopathy associated with ES needs to be differentiated from more benign epilepsy syndromes, particularly benign familial infantile convulsions (BFIC) and benign myoclonic epilepsy of infancy (BMEI) (110–112). A firm diagnosis is necessary before initiation of therapy because many common medications (specifically corticotropin and vigabatrin [VBG]) carry a higher risk of morbidity or mortality than most commonly used anticonvulsants. Like IS, the seizures associated with BFIC and BMEI present in the first year of life. The seizures of BFIC are usually focal, but the EEG may be normal (111). Myoclonic seizures in BMEI can occur in clusters. The EEG may be normal or show generalized spike-and-wave discharges but not the hypsarrhythmia or multifocal independent spike discharges typically seen in West syndrome. Because patients with ES may not display the distinguishing EEG abnormalities early in the disease, a normal or mildly abnormal record does not rule out West syndrome in the early disease stage, and follow-up EEG studies are essential for clarification. A pattern of normal development prior to seizure onset and continued normal development after seizures start is highly suggestive of benign epilepsy syndromes and not an epileptic encephalopathy with IS. In contrast, children experiencing IS may or may not be normal at seizure onset but will invariably demonstrate either developmental regression or failure to achieve developmental milestones in a timely fashion.
As with all forms of epilepsy, the evaluation begins with the history and physical/neurologic examination. The skin should be examined for evidence of neurocutaneous disorders and the fundi for a cherry-red macula suggestive of a storage or mitochondrial disorder or for chorioretinitis indicating possible transplacental infection. In one study, nearly half of all of the etiologic diagnoses were established or suspected by the historical and physical data. Many diagnoses, however, require confirmatory MRI, and once imaging is complete, approximately 70% of patients will have a confirmed etiologic diagnosis (113). In many cases, expensive and time-consuming laboratory studies can be avoided. In the remaining 30% of cases, an etiology will be established for no more than one-third, leaving about 10% of cases in which a diagnosis is determined by results of lumbar puncture or metabolic or genetic testing. A more recent prospect study found etiologies in 61% of children, 33% had no etiology, and 6% were not fully evaluated. (61)
Neuroimaging
Much of the decrease in unknown cases is a result of the advances in MRI techniques of the past 10 to 15 years. Comparing etiologic categories of IS, Riikonen noted that identification of brain malformations increased from 10% between 1960 and 1977 to nearly 35% between 1977 and 1991 (114). Imaging should be considered essential to the evaluation. MRI is preferred to CT scanning because of the greater sensitivity for brain malformations and is not associated with the risk of radiation exposure. CT, however, can show subtle calcifications caused by transplacental infections. In a study of 86 patients with IS, MRI assigned 91% to a symptomatic etiology, most commonly hypoxic–ischemic encephalopathy (30%) (115) characterized by diffuse atrophy and thinning of the corpus callosum. Delayed myelination in 27% of patients did not appear to be associated with any specific etiology (Table 16.1).
Table 16.1 Magnetic Resonance Imaging Findings Implying a Genetic Etiology
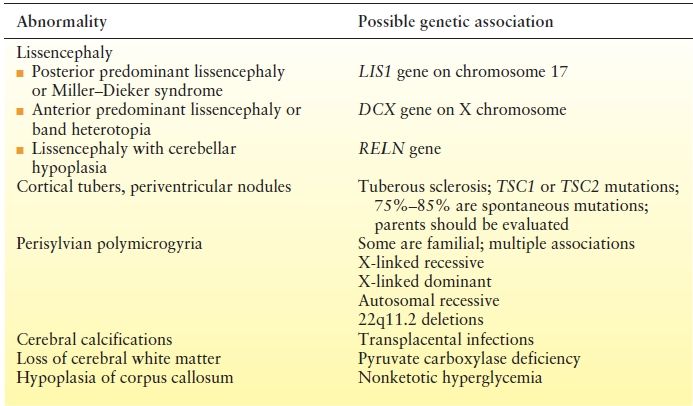
Some MRI abnormalities suggest specific etiologies, many genetically based, which may require further evaluation. Genetic etiologies including Rett syndrome due to various mutations in the MECP2 gene, atypical Rett syndrome due to CDKL5 mutations, and X-linked ARX homeobox gene mutations need to also be considered in the etiologic diagnosis and diagnosed with chromosomal microarrays and specific mutational studies. Additional genetic and metabolic etiologies are continuing to be added to this list as our understanding of the genetic contributions to ES advances (116). Additional associated genetic diagnoses to consider include STXBP1, SCN2A, FOXG1, PCDH19, SLC2A1, ALDH7A1, and POLG. The number of identified genes is growing, and therefore, a complete list is not possible. Improved genetic technology now allows large gene panels to be performed earlier leading to increased genetic diagnosis.
Metabolic Studies
Metabolic studies are indicated to identify more than 50 disorders associated with infantile seizures (117–119). A trial of folinic acid is warranted (120), as is a 100-mg intravenous pyridoxine bolus to exclude pyridoxine-dependent seizures. It is important to remember that further testing is required to completely exclude this diagnosis. Complete blood count, electrolytes (looking for an anion gap), and glucose determinations are appropriate. Measurements of uric acid, transaminases, lactate, pyruvate, ammonia, urine organic acids, and serum amino acids will identify the vast majority of inborn errors of metabolism linked to ES. In the past, phenylketonuria was a relatively common inborn error identified by testing that has been nearly eliminated by neonatal screening. Nevertheless, such screening is not routine in all countries, and measurement of urine amino acid levels will detect phenylketonuria and maple syrup urine disease, as well as other, rarer, metabolic diseases. Zellweger syndrome and neonatal adrenoleukodystrophy are other rare causes of ES that can be diagnosed with the serum very–long-chain fatty acid test (Table 16.2).
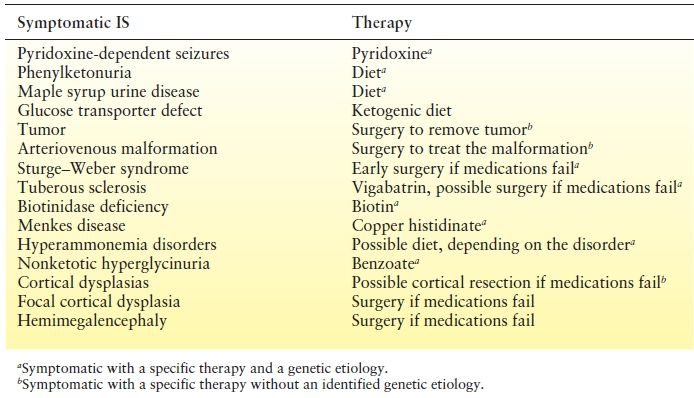
Table 16.2 Etiologies of Infantile Spasms Due to Specific Etiologies That May Respond to Specific Therapies
Lumbar Puncture
A few very rare disorders, such as nonketotic hyperglycinemia, may be detected only by study of the CSF (121). In addition to routine evaluations for glucose, protein, and cells, the CSF should be assessed for amino acids plus lactate and pyruvate to detect possible mitochondrial disorders. CSF neurotransmitters are needed if cerebral folate deficiency is considered as an etiology, and CSF glucose will allow one to discern glucose transporter defects.
PHARMACOLOGIC TREATMENT
Recovery is only considered to have occurred when both the ES and the EEG abnormality of hypsarrhythmia have responded to treatment and ceased. More than 100 years after James West’s initial description of IS, the effectiveness of steroids was first recognized and to date only two medications have class 1 evidence of efficacy: corticotropin and VBG (122,123). Corticotropin and, as of 2009, vigabatrin are approved by the Food and Drug Administration for use in the United States. In addition, some patients respond to valproic acid, lamotrigine, high-dose pyridoxine, topiramate, and zonisamide, while most conventional antiepileptic drugs are ineffective. Some drugs such as carbamazepine or oxcarbazepine may even worsen the seizures, which should be considered when IS are associated with focal features.
Corticotropin and Steroids
The effectiveness of corticotropin and steroids underscores how ES differ from all other epilepsy syndromes. In 1958, Sorel et al. (122) administered 4 to 10 IU/day of corticotropin to seven patients, four of whom responded within a few days while therapy failed in only one patient. In the 45 years since that report, efficacy has been repeatedly confirmed, but agreement is still lacking on the most appropriate dose and duration of treatment. Dosing is complicated by the existence of natural and synthetic forms of corticotropin. Studies of the synthetic product generally used much lower doses than studies of the natural product. It is estimated that 1 IU of synthetic corticotropin is equivalent to 40 IU of natural corticotropin. The synthetic version is used primarily in Japan where a low dose is 0.2 IU/kg/day and a high dose is 1 IU/kg/day. Even with the low dose, 75% of patients responded in one study (124). In contrast, natural corticotropin at doses up to 150 IU/m2 body surface area/day succeeded in 14 of 15 patients; 5 later had relapses for a long-term response rate of 60% (125). A review of seven studies did not confirm a better response with 150 IU/m2/day and 40 IU/m2/day being a common dose (36). The overall long-term response rate ranged from 53% to 91%. Current dosing recommendations are now on the package insert.
Despite its effectiveness for ES, no medication carries a higher potential for significant side effects. Most children develop a Cushing syndrome with obesity, plethora, hypertension, and intense irritability. All patients are at risk for arterial hypertension, electrolyte imbalance, gastric ulcer, growth retardation, cardiomyopathy, or immunosuppression with increased risk of infection. In one study, the risk of serious side effects was 43% with 160 IU/m2/day; the incidence was lower with lower doses (126). Death from infection and cardiomyopathy has ranged from 2% to 5% in some series. Sepsis, tuberculosis, meningoencephalitis, and protracted cytomegalovirus infection have been reported. Corticotropin exacerbates the seizures in a few infants, and treatment for more than a few weeks leads to steroid insufficiency if the drug is stopped abruptly (127). Parents must be fully informed of the associated morbidity and mortality risks (reported at approximately 2% to 5%) before the therapy begins, and these must be balanced against the virtual certainty of mental retardation if the spasms are not rapidly controlled. Careful follow-up with regular measurements of blood pressure, electrolytes, and urinalysis is mandatory. The current risk-to-benefit assessment favors corticotropin, but if another, less hazardous medication proves to be as effective; it would be the drug of choice.
There are few comparative trials of the efficacy of steroids and ACTH but one double-blind trial showed that a 2-week trial of high-dose ACTH was superior to 2 weeks of prednisone, while another found no difference with either compound (128). In addition, some patients may respond to one drug but not the other. Following response to steroids or corticotropin, the relapse rate is high and variably reported between 33% and 56% (35). Relapses usually occur within the first 2 months following the end of treatment, but a second course may give a remission and response in up to 75% of patients (36). Recovery of mental function has been reported between 14% and 58%, particularly in patients with a cryptogenic etiology (125).
Vigabatrin
This drug has been reported to be effective in the treatment of ES of all etiologies, and in patients with TSC, it was superior to steroids (129). In 1991, 29 of 68 patients with medically refractory ES achieved complete resolution with VGB as add-on therapy, as did 12 of 14 patients who had TSC (130). VGB also might be effective in Down syndrome (131).
Since 1997, several controlled trials have been reported. Vigevano and Cilio (132) administered either corticotropin 10 IU/day or VGB 100 to 150 mg/kg/day to children with newly diagnosed IS. Eleven of twenty-three VGB patients responded (one late relapse) compared to 14 of 19 corticotropin patients (six late relapses). Vigabatrin was more effective in patients with TSC or cerebral malformations; corticotropin was more effective in patients with perinatal hypoxic–ischemic encephalopathy. There was no difference in cryptogenic cases. Appleton and colleagues (133) used VGB or placebo for 5 days, followed by open-label VGB. Seven of twenty patients in the VGB group were seizure free at the end of 5 days, as were 2 of 20 in the placebo group. For 14 days, Elterman et al. (134) treated 75 patients with 18 to 36 mg/kg/day (low dose) and 67 patients with 100 to 148 mg/kg/day (high dose) of VGB. Eight low-dose patients and 24 high-dose patients achieved complete control. In a study involving underlying TSC, all 11 patients treated with VGB responded, compared with only 5 of 11 patients treated with hydrocortisone (135). An American Academy of Neurology practice parameter published in 2012 stated that VGB “may be useful for short-term treatment of IS” (136).
VGB appears to be well tolerated. The reports of hypotonia, somnolence, or insomnia (137) are expected in a drug that enhances GABA activity. Constriction of peripheral visual fields, which substantially limits the drug’s use, was not reported until 1997 (138) and now affects from 15% to 50% of patients. In one report (139), constriction occurred in more than 90% of patients who had been taking VGB for a mean of 8.5 years. Foveal function also may be impaired (140). Most studies have suggested that the constriction is not reversible; however, 8 of 12 patients who underwent full withdrawal improved significantly; none of the 12 who continued taking the drug did so (138). The problem is mild enough that most patients are unaware of the disturbance, which becomes apparent only on perimetric studies.
Unfortunately, visual fields are virtually impossible to evaluate in very young children, many with cortical visual impairment and visual inattention unrelated to VGB therapy. Given the catastrophic nature of IS, visual field constriction may be an acceptable price for seizure control and improved opportunity for normal development (139).
VGB has also been reported to be associated with MRI T2 signal abnormalities in the deep grey nuclei (141–143). These characteristic changes involve the basal ganglia, thalamus, dentate nucleus, brainstem, and cerebellum. Restricted diffusion may also be seen with diffusion-weighted imaging and apparent diffusion coefficient maps. Changes appear to resolve with discontinuation of medication, reduction of dose and while medication has continued. Age and dose of VGB appear to risk factors for MRI changes. No clear clinical changes have been associated with reported MRI changes. It is thought that this likely correlates to intramyelinic edema that has been seen in animal models (144,145).
Valproic Acid
Before the availability of VGB around the world, single reports of seizure response to valproic acid were reported with tolerable side effects. In a 1981 report, valproic acid produced an “excellent” response in 4 of 18 patients treated with 20 mg/kg/day (146). A year later, 7 of 19 patients achieved good control (11 had also been treated with corticotropin) (147). Seizures were controlled in 11 of 22 patients treated with up to 100 mg/kg/day for 4 weeks (148). Other patients later responded but also received dexamethasone or carbamazepine, so the effect of valproic acid was less clear. Prospective randomized studies of efficacy against IS are lacking. Because liver failure is a risk in children younger than 2 years of age (149), although none of the reported patients were affected, valproic acid should be used cautiously and probably not as a first line of therapy for IS, particularly if metabolic etiologies have not been excluded (150).
Pyridoxine
A trial of 100 mg of pyridoxine intravenously is appropriate for patients with an unclear etiologic diagnosis because pyridoxine (vitamin B6) dependence can be a rare but highly treatable cause of ES (151). Seizures caused by pyridoxine-dependent epilepsy typically cease immediately following intravenous administration of vitamin B6 but may take several days. As early as 1968, however, it has been known that long-term oral administration of high doses of pyridoxine has been effective against non–pyridoxine-dependent seizures (152). In 1993, 5 of 17 patients treated with 100 to 300 mg/kg/day responded within 4 weeks—most within 1 week (153). In Japan, high-dose vitamin B6 has been the drug of choice (153,154), with reported response rates of 10% to 30%. Loss of appetite, irritability, and vomiting are modest compared with the side effects of corticotropin or VGB, but there may be a high risk of gastric hemorrhage. Pyridoxine is not used as frequently outside of Japan, but given its relatively low-risk profile, a 1- to 2-week trial of 100 to 400 mg may be reasonable before or in addition to other therapies.
Nitrazepam
One of the earliest nonsteroid treatments for IS, the benzodiazepines succeed only occasionally but may be useful if more effective therapies have failed (155–157). Nitrazepam administered to 24 children controlled IS in 11, but hypotonia was reported as a significant side effect (158). A multicenter, randomized comparison of corticotropin and nitrazepam demonstrated no statistical difference between the two medications in significantly reducing spasms (159). Side effects with corticotropin were more severe, leading to discontinuation in six patients. Many reports noted an increase in oral secretions and a higher incidence of aspiration and pneumonia with nitrazepam. Several deaths occurred in one series (160). The incidence of mortality was 3.98 deaths per 100 patient-years when young epilepsy patients were taking nitrazepam and 0.26 deaths per 100 patient-years if the medication had been discontinued (161).
Other Antiepileptic Drugs
None of the new anticonvulsants have enough evidence of efficacy to permit a recommendation. The Japanese experience suggests that zonisamide may be effective in about one-third of patients (162), but controlled and comparison trials are lacking. Five of twenty-five patients had a complete clinical and electrographic response to doses ranging from 8 to 32 mg/kg/day; most responses occurred in 1 to 2 weeks (163). Zonisamide was well tolerated, but 20% of the patients in one study experienced anorexia and one patient lost weight (164). If the more than 30% efficacy figures hold up in controlled studies, zonisamide could become a first-line therapy.
Topiramate up to 25 mg/kg/day was effective in 5 of 11 patients with intractable ES (165). In another study, seizures decreased in 43% of 14 patients but worsened in 29%; no patient became seizure free (166). A prospective study with topiramate in doses up to 8 mg/kg/day found that 10 of 40 patients with new-onset ES responded to topiramate measured by seizure freedom (167). Before reports of aplastic anemia, three of four patients with medically intractable IS responded to felbamate as add-on therapy (168). Because aplastic anemia has not been noted in prepubertal patients, felbamate may be as safe as some other drugs and could be recommended if other medications have failed.
Anecdotal evidence, but no prospective controlled trials, supports the efficacy of lamotrigine. One report noted that 25 of 30 patients became seizure free (169). The usual dose is 6 to 10 mg/kg/day; however, three patients in whom VGB and corticotropin had failed responded to <1 mg/kg/day (169). Use of a low dose is important because rash, the major side effect, depends to some extent on how rapidly the dose is increased. The usual recommendation is a slow rise over 2 months to the minimum expected therapeutic dose. Given the need to control IS as soon as possible, this 2-month requirement decreases the therapeutic value of lamotrigine. If the very low dose is effective, however, lamotrigine becomes a fallback drug (170).
KETOGENIC DIET
The ketogenic diet is a decades-old therapy enjoying a resurgence of interest and widespread application. Two retrospective reports of 40 children suggest control of spasms in 20% to 35% of patients with otherwise intractable disease (171,172). An additional retrospective review of initial treatment of ES with ketogenic diet found seizure freedom within 1 month in 8 of 13 infants (62%) although compared to 18 of 20 treated with ACTH (90%). There was a longer lag in improvement of EEG with ketogenic diet (173). A prospective study for treatment of refractory ES found response rates of 35% at 1 month and 25% at 3 months, although additional medication had been added by 3 months (174). Children younger than 1 year of age can achieve ketosis and may benefit from the diet. Despite generally good tolerability, renal stones, gastritis, hyperlipidemia, and gastroesophageal reflux may occur. More recently, there has been a report of 40% seizure freedom (n = 19) with Modified Atkins Diet (175). This option may be more tolerable to some families.
INTRAVENOUS IMMUNOGLOBULIN
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


