Ethosuximide
Tracy A. Glauser
Diego A. Morita
HISTORICAL BACKGROUND
The development of ethosuximide was a response to the need in the 1950s to develop a more effective, safer, and better tolerated anticonvulsant for the treatment of absence seizures (1). Introduced in the 1940s, trimethadione and its analogue paramethadione were the first anticonvulsants to demonstrate efficacy against absence seizures, but they were associated with significant toxicity (2, 3, 4, 5). These toxicity issues spurred the discovery and testing in the 1950s of the succinimide family of anticonvulsants (ethosuximide, methsuximide, and phensuximide) (5). Of the succinimides, ethosuximide had the greatest efficacy and least toxicity when used against absence seizures (5). Because of this combination of efficacy and safety, ethosuximide has been considered as first-line therapy for absence seizures since its introduction in 1958 (6,7).
CHEMISTRY AND MECHANISM OF ACTION
Ethosuximide (2-ethyl-2-methylsuccinimide), with a molecular mass of 141.2, is a chiral compound containing a five-member ring, with two negatively charged carbonyl oxygen atoms with a ring nitrogen between them and one asymmetric carbon atom (8,9) (Fig. 57.1). Its chemical characteristics include a melting point of 64°C to 65°C, a weakly acidic pKa of 9.3, and a partition coefficient of 9 (chloroform-to-water; pH 7) (9). Ethosuximide is freely soluble in ethanol and water (solubility, 190 mg/5 mL) (9). A white crystalline material, ethosuximide is used clinically as a racemate and is commercially available in 250mg capsules or 250 mg/5 mL of syrup (6,8).
The presumed mechanism of action against absence seizures is reduction of low-threshold T-type calcium currents in thalamic neurons (10,11). The spontaneous pacemaker oscillatory activity of thalamocortical neurons involves low-threshold T-type calcium currents (12). These oscillatory currents are considered to be the generators of the 3-Hz spike-and-wave rhythms in patients with absence epilepsy (12). Voltage-dependent blockade of the low-threshold T-type calcium current was demonstrated at clinically relevant ethosuximide concentrations in thalamic neurons isolated from rats and guinea pigs (10,11,13). Ethosuximide does not alter gating of these T-type Ca2+ channels (5,11). Combining these findings, it is proposed that ethosuximide’s effect on low-threshold T-type calcium currents in thalamocortical neurons prevents the “synchronized firing associated with spike-wave discharges” (10).
There is no evidence that ethosuximide exerts an anticonvulsant effect through other common mechanisms of action (e.g., action at voltage-dependent sodium channels or postsynaptic enhancement of γ-aminobutyric acid [GABA] responses) (5,14). Single-dose administration does not alter brain GABA concentrations in mice (15). The anticonvulsant effect of ethosuximide’s other actions in the brain (i.e., effects on brain enzyme physiology, membrane transport processes, and neurotransmitter processes [14]) are unclear. In cortical tissue, at concentrations significantly greater than those used for anticonvulsant effect, ethosuximide inhibits Na+,K+-adenosine triphosphatase (Na+,K+-ATPase) activity (14,16, 17, 18).
PHARMACOKINETICS
Implications of Racemic Mixture
Ethosuximide has always been used clinically as a racemate. It is theoretically possible that the two enantiomers could demonstrate different pharmacokinetic parameters or anticonvulsant effects. In rats, ethosuximide’s disposition is nonstereoselective (9). Chiral gas chromatographic analysis of enantiomer concentrations in plasma samples obtained for routine monitoring, 33 patients
demonstrated that the enantiomer ratio was close to unity, and there was little interindividual variability (19). This implies that the disposition of ethosuximide in humans is nonstereoselective and that measurement of total ethosuximide for therapeutic monitoring is reasonable and appropriate (9,20). A small study (three pregnancies in two women taking ethosuximide) demonstrated that the nonstereoselective disposition was unaffected by pregnancy, placental transfer, or passage into breast milk (20).
demonstrated that the enantiomer ratio was close to unity, and there was little interindividual variability (19). This implies that the disposition of ethosuximide in humans is nonstereoselective and that measurement of total ethosuximide for therapeutic monitoring is reasonable and appropriate (9,20). A small study (three pregnancies in two women taking ethosuximide) demonstrated that the nonstereoselective disposition was unaffected by pregnancy, placental transfer, or passage into breast milk (20).
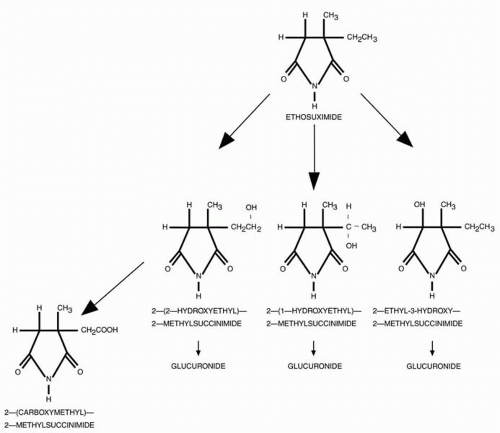 Figure 57.1 Structure and biotransformation pathways of ethosuximide (From Pisani F, Meir B. Ethosuximide: chemistry biotransformation. In: Levy R, Mattson R, Meldrum B, eds. Antiepileptic drugs, 4th ed. New York: Raven Press, 1995:655-658, with permission). |
ABSORPTION
In rats, dogs, and monkeys, absorption is rapid, with nearly complete oral bioavailability in dogs (88% to 95%) and monkeys (93% to 97.5%) (21, 22, 23, 24, 25, 26). In children and adults, absorption is considered to be rapid and nearly complete (90% to 95%), even though no intravenous formulation can be used as a reference standard to determine absolute bioavailability in humans (1,25,27,28). Absorption is reported to remain efficient over multiple administrations (25). In two single-dose capsule administration studies three volunteers given a single 1-g oral dose and four healthy adults given a 0.5-g oral dose, peak ethosuximide plasma concentrations were reached between 1 and 4 hours after administration (26,28,29). A separate study with five institutionalized children that compared capsules and syrup demonstrated peak plasma concentrations within 3 to 7 hours with either formulation (7,25, 26, 27,30). The syrup had a faster absorption rate than the capsules, but the two formulations were bioequivalent (6,7,25, 26, 27,30).
DISTRIBUTION
Tissue Distribution
In rats, ethosuximide distributes evenly to brain, plasma, and other tissues, except for adipose tissue (in which steady-state concentrations are approximately one third of those reached in plasma) (25). Ethosuximide crosses the placenta in rats (26,31), and in dog and rat studies readily passed through the blood-brain barrier (21,26). In dogs, the plasma to cerebrospinal fluid (CSF) ratio was 1.01 ± 0.15, with an estimated half-life of entry into the CSF at about 4 to 5 minutes (21,25,26,32). In one study in rats, the whole-brain to plasma ethosuximide concentrations ratio was near unity, whereas a second study in rats found uniform distribution in four discrete brain areas (cerebral cortex, cerebellum, midbrain, and pons medulla) (24,26). However, a third study in rats receiving a single intraperitoneal dose of 50 mg/kg found a decrease in brain to plasma concentrations over time, suggesting that ethosuximide may be actively transported out of the rat brain (25,33).
In humans, ethosuximide homogeneously distributes throughout the body (6). Saliva, tears, and CSF concentrations are similar to plasma concentrations (25,26,34, 35, 36, 37, 38, 39). In three studies (involving 6, 15, and 19 patients), the respective correlations between saliva and serum concentrations were R = 0.99, R = 0.99, and R = 0.74 (37, 38, 39). A fourth study, which examined concentrations in paired parotid saliva and plasma samples from 10 patients, showed the average saliva to plasma ratio to be 1.04, which appeared constant over the measured time intervals (36). In light of these results, multiple studies have concluded that saliva can be used in lieu of plasma for therapeutic monitoring of ethosuximide (26,34,36, 37, 38,40).
Ethosuximide crosses the placenta in humans and has been detected in cord serum and amniotic fluid at concentrations of 104% and 111% of maternal serum concentrations, respectively (6,41). In two separate reports, ethosuximide was detected in either the urine or plasma of a newborn of a woman receiving long-term therapy (26,42, 43). The serum concentration in the newborn was similar to that in the mother (25,43). Ethosuximide is excreted in the breast milk of mothers receiving long-term therapy (25). In multiple studies, the average breast milk to maternal serum concentration ratio ranged from 0.8 to 0.94 (25,43, 44, 45, 46). The ethosuximide serum concentration of breast-feeding infants of mothers given long-term therapy was 30% to 50% of their mother’s serum concentration (25,45,46). The American Academy of Pediatrics, however, considers ethosuximide to be usually compatible with breast-feeding (47).
Volume of Distribution and Protein Building
The apparent volume of distribution in rats, dogs, and rhesus monkeys ranges from 0.7 to 0.8 L/kg (21,22,25,48). In humans, ethosuximide’s apparent volume of distribution is 0.62 to 0.65 L/kg in adults and 0.69 L/kg in children, implying distribution through total body water (6,25,27,28,35).
METABOLISM AND EXCRETION
Animals
Metabolism is the main method of ethosuximide elimination in animals. In rhesus monkeys and rats, the drug and its metabolites are excreted predominantly by the kidney, with only a small proportion recovered in the feces (25,31). Unchanged ethosuximide accounts for only 12% of urinary recovery in rats (50).
In rats, biotransformation is catalyzed predominantly by hepatic cytochrome P450 (CYP)3A isoenzymes, with possible minor contributions by CYP2E, CYP2B, and CYP2C isoenzymes (9,25,48,51, 52, 53). These CYP enzymes are inducible, and autoinduction has been reported in rats (25,48). The major metabolite in rats and monkeys is 2-(1-hydroxyethyl)-2-methylsuccinimide, and the two minor metabolites are 2-ethyl-3-hydroxy-2-methyl-succinimide and 2-(2-hydroxyethyl)-2-methylsuccinimide (25,54). Ethosuximide provided complete protection against pentylenetetrazol-induced clonic seizures in mice at a dose of 125 mg/kg; in contrast, the major metabolite demonstrated “no significant anticonvulsant activity” (54).
Elimination appears to follow first-order kinetics in animals, except in dogs, in which Michaelis-Menten kinetics may apply (21,25,54). Studies of single- and multiple-dose ethosuximide administration in monkeys have demonstrated comparable elimination half-life and total body clearance (6,22,23). In animals, elimination half-lives range from 1 hour in mice to 9 to 26 hours in rats and 11 to 25 hours in dogs (21,25,54). Steady-state plasma concentrations are significantly higher in the morning than in the evening in rhesus monkeys receiving intravenous ethosuximide at a constant rate. These fluctuations may result from circadian changes in ethosuximide metabolizing enzymes (25,54,55).
Humans
As in animals, metabolism is the main method of ethosuximide elimination in humans. Ethosuximide undergoes extensive hepatic oxidative biotransformation (80% to 90%) to pharmacologically inactive metabolites. Although most of the remaining drug is excreted unchanged in the urine, small amounts of unchanged ethosuximide can be recovered from bile and feces (56). Ethosuximide oxidation is catalyzed mainly by enzymes of the CYP3A subfamily (6). In vitro studies with humanized heterologous CYP microsomal systems showed that ethosuximide is primarily oxidized by CYP3A4, with CYP2E1 playing a minor role in its metabolism (57).
The major metabolite recovered from human urine in patients receiving ethosuximide is 2-(1-hydroxyethyl)-2-methylsuccinimide, of which at least 40% is excreted as a glucuronide conjugate (9,57). Two other metabolites recovered (often as a glucuronide conjugate) from human urine, are 2-ethyl-3-hydroxy-2-methylsuccinimide and 2-(2-hydroxyethyl)-2-methylsuccinimide. The latter metabolite can undergo subsequent metabolism by the hepatic mixed-function oxidase system to form the fourth major metabolite, 2-carboxymethyl-2-methylsuccinimide (9,54, 57) (Fig. 57.1).
In humans, ethosuximide’s elimination follows first-order kinetics. Total-body clearance in adults averages 0.01 L/kg per hour (28) and in two children was 0.016 and 0.013 L/kg per hour (27). This is significantly lower than hepatic plasma flow (0.9 L/kg per hour) implying that ethosuximide does not undergo a significant first-pass effect, and that drug clearance is not blood flow limited (25,26). Total-body clearance has been reported to decrease slightly after repeated dosing (25). Ethosuximide does not induce hepatic microsomal CYP enzymes or the uridine diphosphate glucuronosyltransferase (UDPGT) system (49,58,59). In humans, in contrast to rats, autoinduction does not occur (59,60).
In general, ethosuximide has a long elimination half-life that varies with age. Its mean half-life in adults reportedly ranges from 40 to 60 hours, compared with 30 to 40 hours in children (27, 28, 29,35,54,60, 61, 62, 63). Large variations have been observed in pediatric studies, with half-lives ranging from 15 to 68 hours (27,54,61). In neonates, half-lives ranging from 32 to 41 hours have been reported (43,45). The time to reach steady-state concentration after a dosage change is 6 to 7 days for children and 12 days for adults (6,64). Ethosuximide clearance is reported to be lower in women than men (65). Dose size and repeated dosing do not affect the elimination half-life (35,61).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


