INTRODUCTION
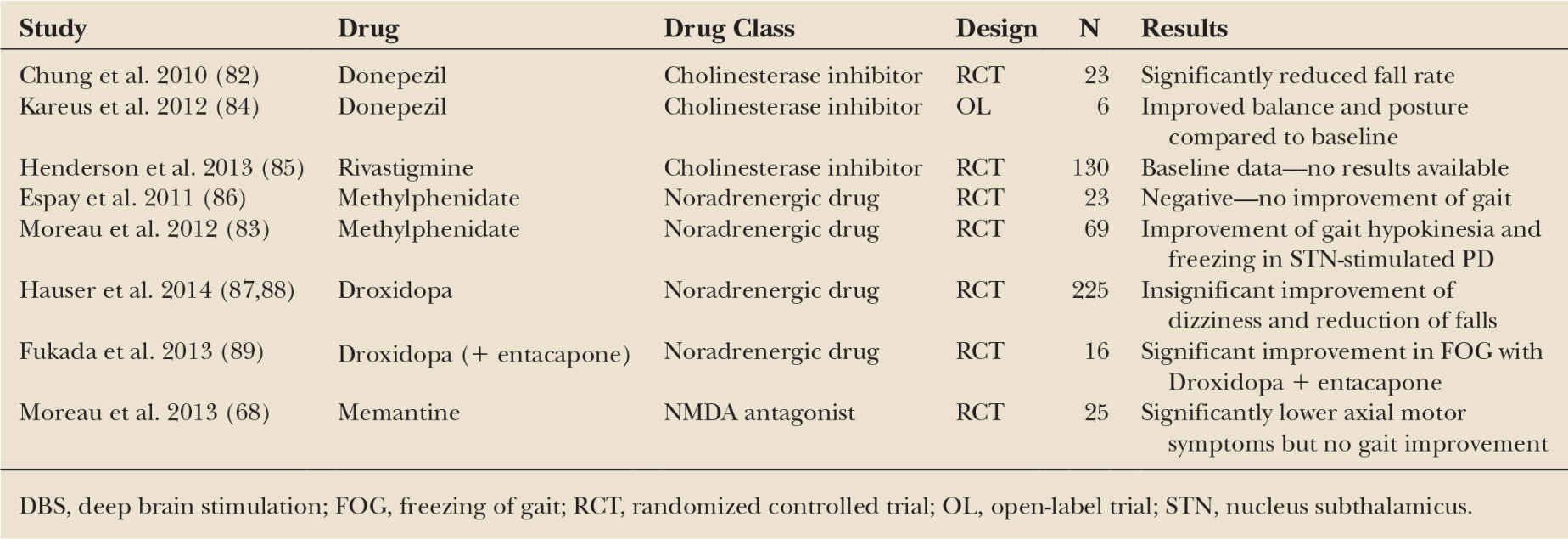
Parkinson’s disease (PD) remains the only neurodegenerative disorder for which there are highly efficacious symptomatic therapies, most importantly dopamine (DA) replacement strategies, which in most patients provide satisfactory motor control for many years. Even after decades of clinical use, levodopa still stands out as the gold standard of symptomatic efficacy, although its long-term use is associated with the development of potentially disabling motor complications. Although a number of pharmacologic therapies are available to reduce levodopa-related motor fluctuations, currently only one drug is approved for clinical use to treat drug-induced dyskinesia in PD (4). Also, as PD progresses, many patients develop motor disabilities that are poorly or inconsistently responsive to dopaminergic drugs, including postural deformities and gait and balance problems. In addition, nonmotor symptoms (NMS) become increasingly common and disabling with advancing PD, and there are currently only few interventions for these with established efficacy through randomized controlled trials (RCTs) (3). Finally, current therapies for PD do not seem to alter the underlying progression of disease, and disease-modifying interventions are a major unmet need (5). In recent years, the focus of clinical trials of PD drugs has reflected these areas of medical need, with growing numbers of studies targeting motor complications from levodopa, NMS, and disease modification (Fig. 11.1).
This chapter will give an overview of potential therapies for PD that are currently in clinical development and testing. These include novel approaches to dopaminergic drug delivery, novel dopaminergic and nondopaminergic agents for motor and NMS, gene- and cell-based therapies, and neuroprotective or disease-modifying strategies.
NOVEL APPROACHES TO DOPAMINERGIC DRUG DELIVERY IN PD
Converging evidence from experimental and clinical studies suggests that discontinuous drug delivery is a major factor for the development of levodopa-related response oscillations and levodopa-induced dyskinesia (LID) (6). Several new approaches to improve dopaminergic drug delivery have reached an advanced stage of clinical development.
LEVODOPA/CARBIDOPA INTESTINAL GEL (DUODOPA)
Initial reports on the efficacy of continuous levodopa delivery as a strategy to smooth out motor fluctuations date back to the 1980s, when several groups were able to show dramatic effects of constant-rate intravenous infusions of levodopa solutions in patients with advanced PD (7,8). These were proof of principle studies, where the need for central lines and the poor solubility of levodopa precluded broad clinical application. Kurlan and coworkers (9) were the first to report about similar effects using intraduodenal delivery of levodopa solutions via nasogastric tubes. Importantly, later studies of intraduodenal levodopa infusions demonstrated not only relatively constant plasma levodopa levels and reduced motor fluctuations but also reductions of preexisting LID (10,11). Again, these studies required the use of large volumes of fluid for infusions due to the poor water solubility of levodopa. Stocchi et al. (12) used infusions of levodopa ethylester, which requires smaller volumes of fluid for solution and was able to show similar results, including reductions of drug-induced dyskinesias.
In recent years, a novel formulation of infusible levodopa has become available in the EU in which the drug is embedded in a carboxymethylcellulose gel, providing a concentration of levodopa/carbidopa of 2 g/0.5 g in only 100 mL. A 100-mL cassette thus contains 2 g of levodopa, allowing for full-day coverage. This novel delivery system uses portable pumps that have programmable delivery rates for amounts between 10 and 2,000 mg of levodopa per hour and delivery times of up to 24 hours. Intrajejunal delivery is achieved through a percutaneous endoscopic gastrostomy (PEG) tube the tip of which is positioned below the Treitz band in the proximal jejunum. Several short- and longer-duration open-label studies have consistently reported marked reductions in daily off-time as well as reduced severity of preexisting LID when such intrajejunal infusion systems were used in patients with fluctuating PD (13–15). One randomized crossover trial comparing levodopa intestinal infusions with oral levodopa/carbidopa over 3 weeks showed marked reductions in plasma levodopa concentration variations and significant increases in on-time without troublesome dyskinesias as compared to traditional oral levodopa administration (13). Recently, levodopa–carbidopa intestinal gel infusion was tested in a randomized controlled, double-blind, double-dummy 12-week trial (16). All 71 patients underwent initial gastrostomy and were then randomized to levodopa–carbidopa intestinal gel infusion plus oral placebo (n = 35) or placebo intestinal gel infusion plus immediate-release oral levodopa–carbidopa (n = 31). This was followed by a 4-week optimization period, during which dosing for patients in either group could be adjusted, and a final 8-week maintenance period. From baseline to 12 weeks, mean off-time decreased by 4.0 hours in the verum group compared with a decrease of 2.1 hours in the immediate-release oral levodopa–carbidopa group, resulting in a significant 1.9-hour group difference (p = 0.002). Likewise, a significant 1.9-hour greater gain in on-time without troublesome dyskinesia was seen in the intestinal gel infusion group compared to the oral levodopa–carbidopa group (p = 0.006). Off-time reduction in this trial was associated with improved quality of life. Overall, 97% of patients experienced an adverse event and 89% of patients had device-related complications, with no difference between groups. Remarkably, the magnitude of off-time decrease over baseline exceeds that reported for any other oral PD treatment so far and is in the order observed in studies of deep brain stimulation. Unfortunately, this trial was not designed to address the issue of reducing LID; so this effect reported from open-label studies still awaits proof from properly designed RCTs. When considering the use of intrajejunal levodopa/carbidopa gel infusions to treat advanced PD patients with motor complications from chronic oral levodopa, physicians should carefully weigh benefits and risks. While robustly effective in reducing motor fluctuations, this treatment is nevertheless invasive and associated with all the risks of PEG such as wound infection, peritonitis, and intestinal bleeding. Teams of movement disorder neurologists and gastroenterologists familiar with key issues around advanced PD seem the best guarantors of optimal results and safest use of this approach.
EXTENDED-RELEASE ORAL FORMULATIONS OF LEVODOPA/CARBIDOPA
Currently available extended-release formulations of levodopa use pharmacokinetic principles that are vulnerable to erratic gastric emptying and incomplete and variable intestinal absorption and have therefore limited efficacy (17–21).
IPX066 is a novel extended-release oral formulation consisting of capsules containing beads of carbidopa and levodopa that dissolve at various rates in the gastrointestinal tract, leading to peak levodopa plasma concentration (Cmax) after 2 hours and sustained therapeutic plasma levodopa concentrations, which remain above 50% of Cmax for around 4 hours (22). An open-label phase II trial in patients with advanced PD showed greater off-time reduction versus standard levodopa/carbidopa (22), a result that was recently replicated in a phase III randomized double-blind double-dummy trial including 393 patients with fluctuating PD with a minimum of 2.5 hours daily off-time (23). Following open-label dose optimization for both IPX066 and standard levodopa/carbidopa, there was 1.2-hour gain in on-time without troublesome LID in the IPX066 group compared to the conventional levodopa/carbidopa group during 13 weeks in the double-blind double-dummy period. Dose frequency was less for IPX066 (3.6 versus 5.1 per day); however, the average total daily dose of levodopa was twice as high in the IPX066 group (1,630 mg versus 815 mg in the conventional levodopa/carbidopa group), which may reflect the lower bioavailability of IPX066–levodopa amounting to 75% compared to standard levodopa/carbidopa preparations (22). Another study has compared IPX066 with combined therapy of levodopa plus the catechol-O-methyltransferase (COMT)-inhibitor entacapone and has also reported superiority of the experimental compound (24). A crucial question is whether IPX066, when used as initial monotherapy, would be associated with a reduced incidence of motor complications compared to standard levodopa. Such comparisons are lacking, although dyskinesia rates have been between 2% and 5% after 30 weeks of treatment with different doses of IPX066 treatment in a double-blind placebo-controlled study (25). After an additional 9 months of open-label follow-up with individualized treatment regimens (mean dosing frequency: three times per day; mean levodopa daily dose: 700 mg), 1.9% of patients on IPX066 developed dyskinesias (26).
Another formulation of levodopa currently undergoing clinical trials is XP21279, a sustained-release prodrug of levodopa that is actively absorbed by a high-capacity natural nutrient transport mechanism located throughout the length of the gastrointestinal tract with subsequent rapid conversion to levodopa. In one study involving 10 PD patients with motor fluctuations, XP21279 was associated with significantly less variability in levodopa concentration compared with standard levodopa indicative of improved pharmacokinetic profile (27). A recent phase II randomized double-blind double-dummy trial failed to show superiority of XP21279 in reducing daily off-time in 28 advanced PD patients compared to immediate-release carbidopa–levodopa in the primary analysis (28). When eliminating statistical outliers (n = 4), however, XP21279 was associated with a significant gain in on-time without troublesome dyskinesia. In addition, there was reduced variability in levodopa plasma concentrations in pharmacokinetic substudy of this trial.
An additional novel extended-release approach for levodopa/carbidopa has been termed the “Accordion pill,” consisting of multiple biodegradable film layers that are folded into an accordion shape and filled into standard oral capsules. Capsules dissolve in the stomach, where films unfold and are retained for extended periods, releasing levodopa/carbidopa for up to 8 hours. Sustained therapeutic plasma drug levels have been attained with twice-daily dosing in a recent phase II study (29).
TRANSDERMAL AND SUBCUTANEOUS DELIVERY OF LEVODOPA
Transdermal patch delivery of levodopa has been attempted through the use of levodopa ethylester, but this approach was not further pursued due to application-site reactions (30). ND0611 is another formulation of levodopa which can be administered by patch application, and preliminary results from phase I studies suggest that this approach can lead to sustained levels of levodopa over 24 hours (30).
ND0612 is a soluble levodopa/carbidopa formulation that can be administered subcutaneously using external minipumps. Phase I results in healthy volunteers show stable levodopa/carbidopa plasma levels over 24-hour infusion cycles, with peak concentrations of around 500 ng/mL at the highest infusion rate (31). A phase II trial in patients with fluctuating PD has recently been initiated (NCT01883505).
INTRAPULMONARY DOPAMINERGIC DRUG DELIVERY
Apomorphine is the only DA agonist with similar effect size on the motor symptoms of PD as the gold standard drug levodopa. It is also the first of its class for which a dry powder formulation has been developed that can be delivered via oral inhalation. Drug inhalation provides ultrarapid access to the systemic circulation via the lung’s large alveolar surface. Inhaled apomorphine has been shown to produce peak plasma drug levels with a tmax of 1 to 3 minutes. This would make this approach attractive for ultrafast off-period reversal in patients with fluctuating PD. Three phase II trials are reported by the same group of authors; a double-blind placebo-controlled dose-ranging multicenter study found rapid apomorphine absorption and reversal from the off-state after 10 minutes, lasting for more than 30 minutes (32). After identifying the ideal dose in a hospital setting, another study reported a significantly higher proportion of off-episodes reversal and insignificantly less total off-time as assessed with patients’ diaries in a home-based setting (33). Using lower doses of inhaled apomorphine, a third study failed to show significant differences compared with placebo (34).
CVT-301 is a formulation of levodopa-loaded aerosol particles suitable for intrapulmonary delivery via a breath-actuated inhaler system which has been shown to result in rapid plasma drug level peaks in less than 20 minutes in healthy, adult subjects (35). A phase II trial in advanced PD patients showed that inhaled CVT-301 rapidly augments levodopa plasma level and results in rapid off-episode reversal in advanced PD patients (36).
NOVEL DOPAMINERGIC DRUGS
Several dopaminergic drugs in development are based on the principle of enzyme inhibition in order to enhance central dopaminergic transmission. These include novel inhibitors of monoamine oxidase type B (MAO-B) and COMT as well as an approach increasing the ratio of the currently used peripheral amino acid decarboxylase (AADC)-inhibitor carbidopa to levodopa.
NOVEL MAO-B INHIBITORS
Safinamide is a novel reversible MAO-B inhibitor with additional mechanisms of action, including glutamate-release inhibition and sodium channel–blocking properties. In a randomized, placebo-controlled study of 172 early PD patients, safinamide treatment was associated with significantly greater percentages of responders (defined by ≥30% improvement in UPDRS motor score) compared to placebo after 3 months: 30.9% on low-dose (median 40 mg/day) and 37.5% on high-dose (median 70 mg/day) safinamide compared to 21.4% of patients receiving placebo (37). A further placebo-controlled 24-week trial of adjunct safinamide included 269 early PD patients receiving a stable dose of a single DA agonist (38). While the lower dose (100 mg/day) used in this trial was associated with significant improvements on UPDRS motor scores (−6.0 points compared to −3.6 points on placebo), the difference between the 200-mg/day dose and placebo failed to reach statistical significance. A 12-month blinded extension study failed to show significant differences in the median time from baseline to additional drug intervention (increase in DA agonist dose; addition of another DA agonist or levodopa) between the pooled data from the safinamide groups compared to placebo (39). A post hoc analysis revealed a significantly lower rate of intervention in the 100-mg/day arm compared with placebo (25% versus 51%, respectively), suggesting that at this dose safinamide may be effective as add-on treatment to DA agonists in early PD patients also in the long term. This was confirmed in a large phase III 24-week RCT (MOTION) in 607 early PD patients, where safinamide 100 mg/day met the primary objective of significantly improving motor symptoms and quality of life compared to placebo as add-on to a single DA agonist (40). A recent placebo-controlled, double-blind trial in 669 PD patients with levodopa-associated response oscillations showed significant increases in daily on-time with both 50 mg and 100 mg of safinamide per day as compared to placebo, with a main increase of 1.4 hours of on-time per day as compared to 1.0 hours on placebo (41). In addition, there was a significant gain in on-function, as assessed by the motor section of the UPDRS (−6.1 and −6.9 score points on 50 and 100 mg of safinamide compared to −4.3 points on placebo). An open-label, two-year extension study provided some evidence for potential reduction in dyskinesia severity with 100 mg of safinamide (42). The latter findings were confirmed in a large phase III 24-week RCT (SETTLE) in 484 fluctuating PD patients, where safinamide 50 to 100 mg/day met the primary efficacy objective of significantly improving on-time without any increase in troublesome dyskinesia (43).
NOVEL COMT INHIBITORS
The nitrocatechol compound entacapone is currently the only COMT inhibitor available as first-line treatment for patients with levodopa-related motor response fluctuations. Two novel nitrocatechol compounds have recently entered clinical development.
Nebicapone (BIA 3-202) was assessed in a phase IIb placebo- and active-controlled randomized study in 252 PD patients with levodopa-induced motor fluctuations and was associated with a significant reduction in off-time versus placebo and comparable to the active comparator entacapone. However, these effects were only significant at the highest doses tested, which also led to clinically relevant liver enzyme elevation in 4 out of 46 patients, such that further clinical development was not pursued (44).
Obicapone (BIA 9-1067) is another highly potent COMT inhibitor (45). The drug has an interesting pharmacokinetic profile with long half-life and a potential for once-daily dosing. Phase II studies have shown significant increases in on-time. A randomized double-blind phase III trial comparing 25 and 50 mg of once-daily obicapone to placebo in 407 patients with fluctuating PD (BIPARK II) has recently been completed and shown significantly reduced daily off-time of −1.7 hours (versus −1.1 hours on placebo) for the 50-mg arm (46). The BIPARK I trial of obicapone includes an entacapone active comparator arm, but results are not yet available (NCT01568073).
CHANGING THE LEVODOPA/AADC INHIBITOR RATIO
Inhibiting peripheral AADC with carbidopa or benserazide increases bioavailability and, to a lesser extent, plasma half-life of levodopa, and current formulations of the drug come in fixed combinations of levodopa plus AADC inhibitor using ratios of 4:1, although there is also a 10:1 formulation of levodopa/carbidopa (4). Increasing the dose of carbidopa beyond the currently used level of 25 or 50 mg per 100 or 200 mg of levodopa has been shown to further extend t1/2, Cmax, and AUC, and a recent crossover double-blind trial has compared clinical effects on off-time in patients with fluctuating PD when using standard tablets of 75 mg/200 mg to 150 mg/200 mg levodopa/entacapone containing between 18.75 and 37.5 mg of carbidopa (= standard LCE tablets) versus corresponding levodopa/entacapone dose formulations containing fixed amounts of 65 or 105 mg of carbidopa per tablet irrespective of levodopa dose (47). The 65-mg carbidopa dose arm showed significantly greater reduction in daily off-time compared to standard LCE in the primary analysis, and both the 65- and the 105-mg dose were superior to standard LCE on this outcome in a carry-over–adjusted analysis. Although potentially clinically useful, this new formulation of levodopa may not enter full clinical development due to patent and ultimately reimbursement issues.
NONDOPAMINERGIC DRUGS IN DEVELOPMENT
For many years, research in novel pharmacologic therapies for PD has focused on the dopaminergic system. However, nondopaminergic neurotransmitters have increasingly been recognized as potent modulators of basal ganglia circuit activity (48,49). Drugs targeting these nondopaminergic mechanisms may have antiparkinsonian activity as monotherapy, modulate the effects of levodopa to reduce response oscillations or dyskinesia, or even affect levodopa-unresponsive motor and NMS. Fig. 11.2 gives an overview of the potentially involved nondopaminergic neurotransmitter systems.
ADENOSINE A2A-ANTAGONISTS
Of the four adenosine receptors, the A2A subtype is highly expressed in the basal ganglia, especially the striatum. It colocalizes with DA D2 receptors on medium spiny neurons projecting to the external segment of the globus pallidus (GPe) as part of the indirect pathway. Adenosine A2A receptors act as modulators of the D2 receptors, and in PD, their activation contributes to overactivity of the indirect pathway. Therefore, A2A antagonists may reduce striatopallidal overactivity and ameliorate parkinsonism. Several candidates of this class of agents have progressed to different stages of clinical development.
Istradefylline, the first A2A antagonist evaluated in proof-of-concept studies and later in phase II trials, showed positive signal for reduced response oscillations when given to patients with fluctuating PD (50–53). Two subsequent randomized controlled phase III trials showed off-time reduction of 0.7 hours with 20 mg in one trial (54) and 0.65 hours with 20 mg and 0.92 with 40 mg istradefylline per day in the other trial (55). A third large randomized controlled phase III study in 610 PD patients on levodopa therapy with motor response complications failed to detect significant differences in daily off-time with 10-, 20-, and 40-mg istradefylline per day (56). However, a meta-analysis of all available randomized trials up to 2012 found that istradefylline significantly reduces off-time and diskinesias and increases UPDRS III scores in the on state at 40 mg per day (57). A recent Japanese phase III trial again showed that off-time was significantly reduced in the istradefylline 20- and 40-mg/day groups compared with placebo (58), and the substance has been approved as adjunctive treatment for advanced PD patients in Japan. Istradefylline has also been studied as a monotherapy in 176 early PD patients, but at 40 mg/day the drug had no beneficial effect in improving motor symptoms compared to placebo (59). A phase III trial in patients with fluctuating PD is currently ongoing in the European Union.
Preladenant is a potent and selective competitive antagonist of the A2A receptors. A recent phase II trial in 253 advanced PD patients with motor fluctuations showed significant reductions in off-time (−1.0 and −1.2 hours) and increased daily on-time with nontroublesome dyskinesias at the 5- and 10-mg doses (60). In an open-label follow-up study, preladenant 5 mg twice daily was associated with off-time reductions of 1.4 to 1.9 hours/day relative to the baseline of the double-blind study over 36 weeks (61). However, three separate phase III trials of preladenant in early PD patients and advanced PD patients versus placebo and versus rasagiline have been completed and initial review of the data has failed to provide evidence of superiority, such that further development was discontinued (Merck News Release, May 2013).
Tozadenant (SYN115) is a member of novel nonxanthine–nonfuran A2A antagonists exhibiting a 100-fold selectivity for A2A versus other adenosine receptors. It has recently been tested in a phase IIb trial in 420 PD patients with wearing-off fluctuations over 12 weeks, who were randomized to 60-, 120-, 180-, or 240-mg b.i.d. tozadenant, or matching placebo (62). Significant reductions in placebo-corrected changes from baseline in off-time (−1.1 and −1.2 hours) and in mean placebo-corrected UPDRS III scores (−2.2 and −2.5) were observed with the 120- and 180-mg doses, while troublesome dyskinesia were not significantly increased. A previous proof-of-concept functional MR imaging study was able to demonstrate reduced activity of the indirect pathway following administration of SYN115, consistent with A2A antagonism (63).
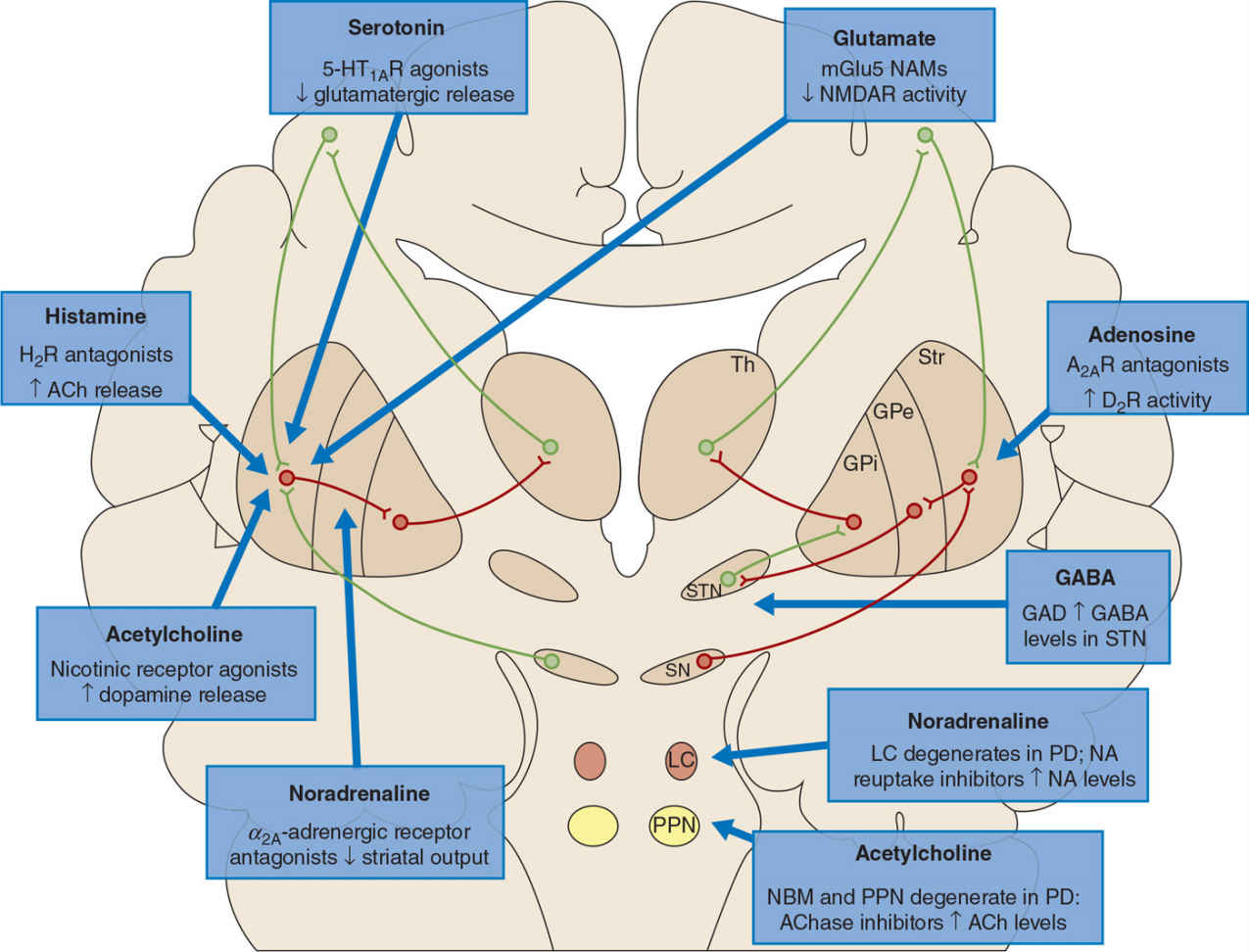
Figure 11.2. Nondopaminergic nuclei affected in PD include the pedunculopontine nucleus (PPN) and nucleus basalis of Meynert (not shown), which produce acetylcholine, as well as the locus coeruleus (LC), which produces noradrenaline. These nuclei project to the basal ganglia as well as other brain regions to supply their respective neurotransmitters. Within the basal ganglia, current drugs that target the acetylcholine, glutamate, histamine, or noradrenaline systems appear to modulate the direct pathway (left), whereas those that target the adenosine and GABA systems may influence the indirect pathway (right). AChase, acetylcholinesterase inhibitor; GPe, globus pallidus externa; GPi, globus pallidus interna; Str, striatum; Th, thalamus. (From Kalia LV, Brotchie JM, Fox SH. Novel nondopaminergic targets for motor features of Parkinson’s disease: review of recent trials. Mov Disord 2013 Feb;28(2):131–144.)
Interestingly, caffeine—a nonselective antagonist at A1 and A2A receptors—was shown to improve objective motor measures of PD in an RCT, while excessive somnolence was not statistically significantly improved (64). A larger long-term trial also aiming to assess potential disease-modifying effects of caffeine is currently ongoing (NCT01738178).
GLUTAMATERGIC ANTAGONISTS
The development of LIDs has been linked to relative overactivity in the direct striatopallidal pathway (49). Glutamate receptors, especially NMDA (N-methyl-D-aspartate), mGluR5 (metabotropic glutamate receptors type 5), and AMPA (amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid) receptors, abundantly expressed in striatal neurons are thought to increase DA D1–mediated activity of the direct pathway of GABAergic striatal neurons (65). Inhibiting glutamatergic excitatory input onto medium spiny projection neurons of the indirect pathway through glutamate-receptor antagonism is therefore a promising target for the development of novel antidyskinetic drugs in PD. Currently, amantadine, a weak NMDA receptor antagonist, remains the only drug with established antidyskinetic efficacy (4,66). An extended-release formulation of amantadine has been developed and positive results have recently been reported in a phase II/III study (67). Memantine, an uncompetitive, partial antagonist at the NMDA receptor approved to treat dementia in Alzheimer’s disease, has recently been tested with regard to axial PD symptoms in 25 advanced PD patients (68). While there was no change in the primary study endpoint (stride length), decreased dyskinesia rating scale scores were observed after 90 days of mementine treatment. In contrast, the AMPA antagonist perampanel has failed to show benefit in reducing off-time and duration or disability of LID in a phase III RCT (69).
Preclinical studies with the mGluR5-negative allosteric modulator mavoglurant (AFQ056) have indeed shown reductions of LID in the MPTP model (70). Two small placebo-controlled phase II trials (34 patients each) have both shown significant reductions in dyskinesias as assessed by the abnormal involuntary movement scale (AIMS), the Lang-Fahn Activities of Daily Living Dyskinesia Scales, or the UPDRS part IV items 32 and 33 (71). A larger phase IIb dose-ranging study included 197 patients with LID treated with five dose levels of AFQ056 (10, 25, 50, 75, and 100 mg twice daily) or placebo for 12 weeks (72). Significant improvements in the primary outcome, modified AIMS, were observed for the 100-mg b.i.d. dose compared to placebo, and the same dose also showed significant reductions in the UPDRS IV item 32 (dyskinesia severity), while other dyskinesia scales (PDYS-26 or SGIC) did not show significant improvements. Adverse event related dropout rates were high in the high-dose arm, including neuropsychiatric side effects like confusion and hallucinations. Two phase IIb trials, one in 63 patients using the immediate-release formulation and one in 140 subjects using a modified-release formulation, have recently been completed and, based on the sponsor’s news release, failed to provide evidence for antidyskinetic efficacy (Novartis news release, November 2013).
Dipraglurant, a negative allosteric modulator of the mGlu5 receptor, showed antidyskinetic effects in MPTP and haloperidol animal models of PD (73,74). A phase II RCT investigating the effects of dipraglurant in advanced PD patients showed a significant reduction in dyskineasia severity as measured with the modified AIMS and a gain in on-time without dyskinesia (75).
α-ADRENERGIC ANTAGONISTS
α-Adrenergic 2A and 2C receptors modulate GABAergic transmission at the level of the striatopallidal projection, which is hyperactive in patients with LID dyskinesias (49).
Fipamezole is an adrenergic α2-receptor antagonist that has shown antidyskinetic activitiy in the MPTP Parkinson monkey model (76). A proof-of-concept study in 21 PD patients with LID used levodopa infusion paradigm to elicit dyskinesias (77). The effects of fipamezole administered as a buccal spray in ascending doses of 30, 60, and 90 mg or placebo were assessed both regarding dyskinesia severity, as well as duration of levodopa response following stop of infusion. Dyskinesia severity decreased by 23% at 60 mg of fipamezole and 31% at 90 mg (p <0.05 versus placebo). In addition, levodopa response duration was significantly prolonged with the 90-mg dose by 41 minutes. A larger, double-blind, placebo-controlled, phase IIb study (FJORD) included 115 subjects in the United States and 64 patients in India (78). Study duration was 4 weeks, and doses were escalated to a maximum of 90 mg three times daily. While there was no significant difference between placebo and active drug for this total study population on the primary endpoint of the LID rating scale, a prespecified subanalysis of US subjects demonstrated a significant dyskinesia reduction in the 90-mg dose.
HISTAMINERGIC ANTAGONISTS
Famotidine, a drug currently available for use in the clinic in a different setting, has been shown to enhance antiparkinsonian action of low-dose levodopa and reduce peak-dose LID in a preclinical testing (79). Antidyskinetic properties of this drug are currently in phase II clinical testing (NCT01937078), while the potential mechanism of action of this class of drugs is unclear.
DRUGS TARGETING GAIT AND BALANCE PROBLEMS OF ADVANCED PD
Motor problems that are poorly responsive to levodopa or other dopaminergic drugs are a major source of disability in advanced PD. They include dysarthria and dysphagia, axial and limb deformities, and disorders of gait and balance. The latter represent a particular area of medical need, since up to 70% of patients with advanced PD report regular falling and 20% are estimated to sustain falls-related fractures (80,81). The mechanisms underlying freezing of gait and other types of gait disorders as well as postural instability in PD are poorly understood but may include defective cholinergic and adrenergic neurotransmission due to cell loss in key brainstem areas like the pedunculopontine nucleus (PPN) and locus coeruleus (LC). As a consequence, recent trials have tested efficacy of cholinergic and adrenergic drugs on gait and balance function in advanced PD patients. For instance, in a RCT in 26 PD patients with more than two falls per week, donepezil significantly reduced falls compared to placebo during this 6 weeks study (82). Another trial showed significant improvements of gait hypokinesia and freezing in STN-stimulated advanced PD patients with methylphenidate compared to placebo (83). Further trials targeting gait and balance problems of advanced PD using cholinergic or adrenergic approaches are listed in Table 11.1.
DRUGS IN DEVELOPMENT FOR NMS
NMS, including disorders of mood and affect, cognitive dysfunction and psychosis, autonomic failure, and disorders of sleep–wake cycle regulation, are now recognized as important elements in the clinical spectrum of PD. They become increasingly prevalent and disabling over the course of the disease such that in advanced stages of the illness NMS may represent the main therapeutic challenge. Despite the high prevalence and associated burden of NMS in PD, evidence for efficacy of interventions from RCTs is lacking for many of these problems, although the number of clinical trials focusing on NMS in PD is on the rise (see Fig. 11.1). Nevertheless, there have only been few trials successfully reaching the final stages of clinical development (3). Droxidopa (L-threo-3,4-dihydroxyphenylserine), an oral norepinephrine prodrug, has recently been approved by the FDA for the treatment of neurogenic orthostatic hypotension (NOH) associated with PD, multiple-system atrophy, and pure autonomic failure (FDA, press release 02/2014). Pimavanserin, a novel selective serotonin 5-HT2A inverse agonist, was recently tested in a phase III RCT in 199 PD patients with psychosis (90). After 4 weeks of treatment, there was a significantly greater reduction of the PD-adapted scale for assessment of positive symptoms (SAPS-PD) scores in the pimavanserin compared to the placebo arm (−5.8 versus −2.7 points), while motor function remained unchanged. Table 11.2 gives an overview of recent and ongoing trials testing potential drugs to treat NMS of PD.
< div class='tao-gold-member'>