HISTORICAL INTRODUCTION AND TERMINOLOGY
Frontotemporal dementia (FTD) is a new name for Pick’s disease (PiD). Pick’s initial case (1) of a progressive aphasic patient with behavioral disturbances had only gross autopsy examination, but the clinical description and its relationship to focal atrophy are the basis of the syndrome. Later, PiD was defined on the basis of round silver staining inclusions (Pick bodies), described by Alzheimer (2), which later turned out to be containing abnormally phosphorylated tau or a tauopathy (3), thereby turning a clinical disease into a pathologic one.
Even though Pick had not included any extrapyramidal symptoms in his series of papers, there have been several case descriptions of PiD where the patients had prominent extrapyramidal features (4–6). Sometimes, unilateral rigidity and parkinsonism were the first symptoms to attract attention. It was also recognized that subcortical changes occur in PiD, even without extrapyramidal symptomatology. The “Akelaitis variety” or type B PiD terminology has fallen into disuse.
Frontotemporal atrophy was demonstrated with increasing frequency in vivo, first with CT and later with MRI and SPECT. Several studies applied new labels such as frontal lobe degeneration (FLD) (7) and subsequently FTD (8). It became apparent that cases of clinical PiD may not show the typical histologic picture of Pick bodies. The use of PiD is now increasingly restricted to pathology. The terms of FLD or FTD were initially applied mainly for the behavioral presentation while reserving the diagnosis of PiD to the postmortem finding of Pick bodies. Since FTD is used for both the overall disease and the behavioral presentation ambiguously, the term “Pick complex” (PC) was suggested to encompass all the related entities, including primary progressive aphasia (PPA) and corticobasal degeneration (CBD) clinically and pathologically (9). Others have promoted “frontotemporal lobar degeneration” (FTLD) (10), and this term is now increasingly used to designate the pathologic entity, including tauopathies (FTLD-T) and ubiquinopathies (FTLD-U), and its subclassifications (FTLD-TDP-43, FTLD-FUS).
The syndrome of axial dystonia, bradykinesia, falls, dysphagia, and vertical gaze palsy was described as progressive supranuclear palsy (PSP) by Steele Richarson and Olszewski. (11), as a unique entity near parkinsonism (“parkinsonism plus”). Bradyphrenia, behavioral abnormality and dementia, was part of the initial description, and PSP became the most representative example of “subcortical dementia,” characterized by “bradyphrenia” or slow responses, better recognition than recall, and the preservation of visuospatial and language function (which later turned out to be inaccurate) (12). However, the recognition of FTD pathology in cases of clinical PSP and vice versa only came later, and there are still many who disregard the relationship. More recently, the clinical syndrome of PSP is abbreviated as PSPs, and the classical presentation has been renamed as “Richardson’s syndrome,” an attempt to separate it from the “parkinsonian” or other presentations of PSP pathology (13).
Rebeiz, Kolodny, and Richardson (14) described corticodentatonigral degeneration and recognized the similarity of the pathology to PiD (silver staining inclusions and ballooned neurons). However, they considered it a new entity, different from the aphasic or behavioral clinical presentation of PiD and stated that “mental faculties were relatively preserved” in their cases. The clinical syndrome of unilateral rigidity, prominent apraxia, reflex myoclonus, and the alien hand syndrome was relabeled CBD 23 years later (15). Case reports with clinical features of CBD but other pathologies such as Pick bodies appeared (16), and cases of pathologically typical of CBD but with a clinical presentation of PPA or FTD without the extrapyramidal features were described. After the initial reports from movement disorder clinics, CBD patients began to appear in cognitive clinics with varied descriptions of cognitive deficits. CBD pathology was confirmed as a substrate for PPA (16–19).
We suggested that the clinical syndrome of CBD consisting of prominent apraxia, asymetric extrapyramidal syndrome, and alien hand phenomenon should be designated as corticobasal degeneration syndrome (CBDS), and CBD should be used for the pathologic picture (19,20). Corticobasal syndrome (CBS) was suggested later and became the currently popular abbreviation for the clinical syndrome. (21) We shall continue to use CBDs for the clinical syndrome, because it parallels the frequently used PSPs, and CBS is less immediately evident as to its meaning.
THE BIOLOGIC OVERLAP OF CBD/PSP AND FTD
There is a complex relationship of FTD to CBD/PSP at many levels. The clinical, pathologic, genetic, and molecular levels of overlap of CBD and PSP are currently well recognized, albeit not without controversy (22–28). Often, both the clinical and pathologic entities are dealt with separately. Even after considering the extensive overlap, it is argued that FTLD, CBD, and PSP should be considered as pathologically similar but distinct syndromes (29). In the recent consensus meeting on CBD and CBDs in Baltimore, there was a great deal of discussion and reluctance to leave out PSP from the compilation of the criteria for CBD (30). PSP and CBD are currently considered pathologic diagnoses, with different clinical phenotypes that overlap and share features with FTD syndromes and Parkinson’s disease (31). The emphasis is on pathologic classification of the syndromes with variable clinical manifestations (see the pathology section below). The actual distribution of the clinical and pathologic diagnosis is dependent on the origin of the material. Cognitive clinics will have predominant CBDs, and movement disorder clinic-associated brain banks with retrospective studies will show more PSP and PSPs diagnosis. If the frequent cognitive features at presentation are not considered as an essential part of the CBDs presentation, then the diagnosis of CBD is made less frequently in vivo (27).
This chapter presents an integrative approach, where molecular biology and longitudinal clinical manifestations are considered together. Evidence is presented that even the tau–nontau dichotomy has so many overlapping features that the condition should be considered as an interrelated biologic entity, rather than a heterogeneous disorder or multiple different diseases.
TAUOPATHIES, CBD/PSP, AND FRONTOTEMPORAL LOBAR DEGENERATION–TAU (FTLD-TAU)
Abnormally aggregated tau proteins are constituents of various forms of degenerative dementia, including Alzheimer’s disease (AD), PiD, CBD, PSP, FTD, the parkinsonism–dementia of Guam, dementia pugilistica, renamed chronic traumatic encephalopathy (CTE), argyrophilic grains disease, sporadic multisystem tauopathy (MST) with globular inclusions, and diffuse neurofibrillary tangle dementia with calcifications (DNTC). Most of these are cortical or subcortical varieties of FTLD–tau. Even AD, which is not considered a primary tauopathy, but an amyloidopathy, may also mimic the FTLD/CBD spectrum. In AD, abnormally phosphorylated tau forms three bands on Western blot studies with molecular weights of 55, 64, and 69 kDa, while PiD has 64-kDa and 55-kDa bands, and CBD and PSP 69- and 64-kDa doublets (31). At times, FTD has the same tau triplet as in AD. Sometimes, different band compositions are obtained from different parts of the brain (32). Normal tau proteins contribute to axonal transport by binding to microtubular protein (31,33). Six tau isoforms are created by the differential splicing of exon 10, making three or four repeats of the microtubular-binding domain of tau. Three-repeat (3R) tau predominates in PiD, and 4R tau is characteristic in CBD and PSP.
PiD as defined pathologically by the round neuronal inclusions or Pick bodies is turning out to be the least common FTLD–tau. PSP and CBD are now considered more common, and these conditions have extensive clinical and pathologic overlap, with no distinctive biomarker that permits their differentiation. Autopsy differentiates globose tangles, oligodendroglial coiled bodies and tufted astrocytes in PSP and threads, ballooned neurons, pretangles, and astrocytic plaques in CBD, but there is overlap of some of these features (22,34,35). There is particularly overlap between subcortical nuclei affected in PiD, CBD, and PSP (22). The anatomical distribution of tau pathology corresponds to the clinical presentation to some extent, but the clinical presentation is only predictive of the pathology to a limited degree.
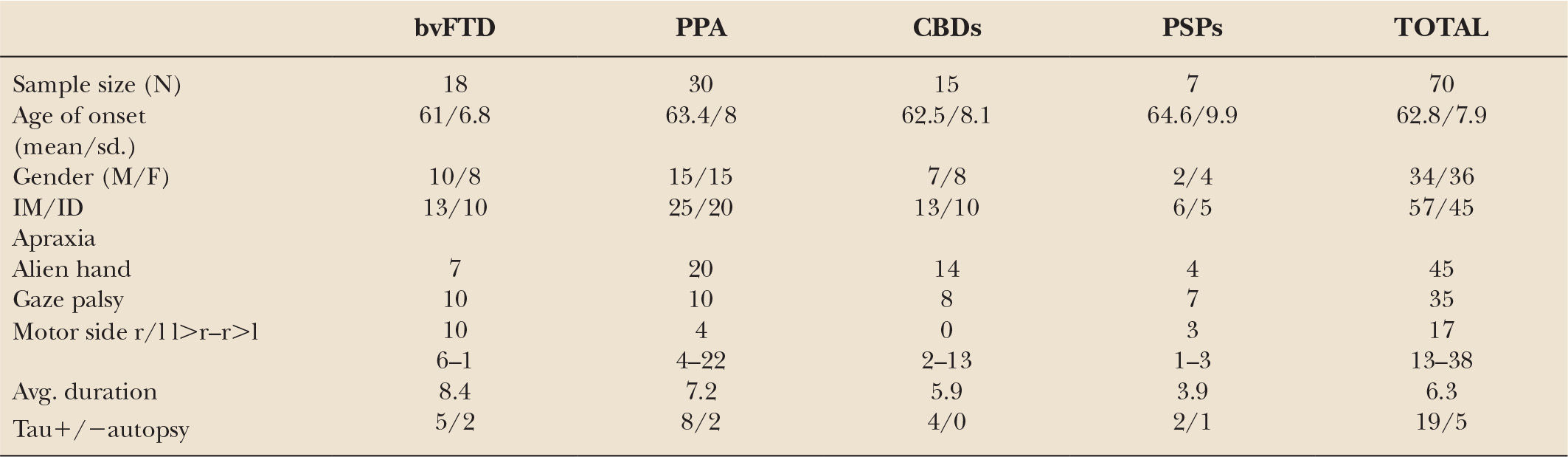
While differences are noted between pathologically diagnosed CBD presenting as CBDs versus PSPs, no clinical features reliably distinguish whether a clinical presentation of PSPS will demonstrate CBD or PSP pathology at autopsy (27,28). A recent clinicopathologic series found that in subjects presenting with PSPs, the only clinical characteristics differentiating subjects with CBD versus PSP pathology were more executive and behavioral abnormalities and more urinary incontinence in the PSPs patients with CBD pathology (28). We recently updated our longitudinal clinical cohort of 319 FTD, with a high autopsy rate, 80 of which were considered clinically CBDs or PSPs sometime during the course of their illness (36). In 24 that came to autopsy, tauopathy was found in 19, features of CBD in 11, PSP in 3, and Pick body disease in 4. In two other cases, features of PSP/CBD were both present. FTLD-U was seen in three, one had Gerstmann–Straussler–Scheinker disease, and one had AD. The evolution of these syndromes and their outcome are summarized in Figs.17.1 to 17.4. The clinical features of the various extrapyramidal syndromes in FTD in our clinic are summarized in Table 17.1 (37).
In a larger pathologic series (179 PSP and 29 CBD), 42% of CBD and 54% PSP autopsies had clinical PSPs (27), renamed as “Richardson’s syndrome” (13). In variants of PSP presenting with focal cortical syndromes, such as FTD, CBS, and apraxia of speech (AOS), there is greater cortical pathology than in typical PSPs. CBDs had greater tau pathology in the primary motor and somatosensory cortices and putamen, while those with PSPs had greater tau pathology in limbic and hindbrain structures (28). Compared with PSP, patients with CBD associated with PSPs had less neuronal loss in the subthalamic nucleus, but more severe neuronal loss in the medial substantia nigra and greater atrophy of the anterior corpus callosum. The results suggested that PSPs can be a clinical presentation of CBD pathology. Atrophy of anterior corpus callosum may be a potential neuroimaging marker to differentiate CBD from PSP in patients with PSPs. In variants of PSP presenting with levodopa responsive parkinsonism, as well as pure akinesia and gait failure (PAGF), there is less cortical pathology and more severe degeneration in the globus pallidus, subthalamic nucleus, and substantia nigra. The results from the Mayo clinic PSP bank with both CBD and PSP pathologies also indicate that the commonest underlying pathology of CBDs is PSP. However, these studies have been likely biased in favor of PSP pathology because they originated from a parkinsonism or PSP brain bank. In contrast, in a clinical series of 545 dementia cases, 45 had CBD, and only 7 had PSP pathology (38), which corresponds with our experience from a cognitive neurology clinic. Although heterogeneity is the usual word applied, it is not known if these tauopathies are variations of the same disease or represent different entities.
FTLD-U AND NONTAU PATHOLOGIES
The overlap between CBD, PSP, and FTD is not confined to tau biology and also extends to the other major pathologic entities of FTD. The ubiquitinated inclusions (FTLD-U) containing transactive response DNA-binding protein with molecular weight of 43 kDa (TDP-43) have been described in association with CBDs and PSPs (38–42). TDP-43 colocalized with tau in 5 of 19 PSP cases (26%) and in 2 of 12 CBD cases (17%) in one of the studies (42). Other transitory syndromes such as FTD with supranuclear palsy and chorea with PSP-like distribution of TDP-43 pathology (43), or CBD with olivopontocerebellar atrophy and TDP-43 pathology have been described (44). We have recently published a family with behavioral variant FTD (bvFTD) and TDP-43 pathology in several members and tau-positive, classical PSP pathology associated with bvFTD and PSPS in the propositus (45). FTLD-FUS (fused in sarcoma) protein abnormality in basophilic inclusion body disease (BIBD) and neuronal intermediate filament inclusion disease (NIFID) have been also observed with subcortical pathology and PSP symptomatology (46). It should be mentioned that a number of CBDs patients turn out to have AD pathology in some studies (47–49), although in our experience this is unusual (36,37). CBDs, especially when parietal features are prominent, is considered less predictive of tau pathology than PSPs.
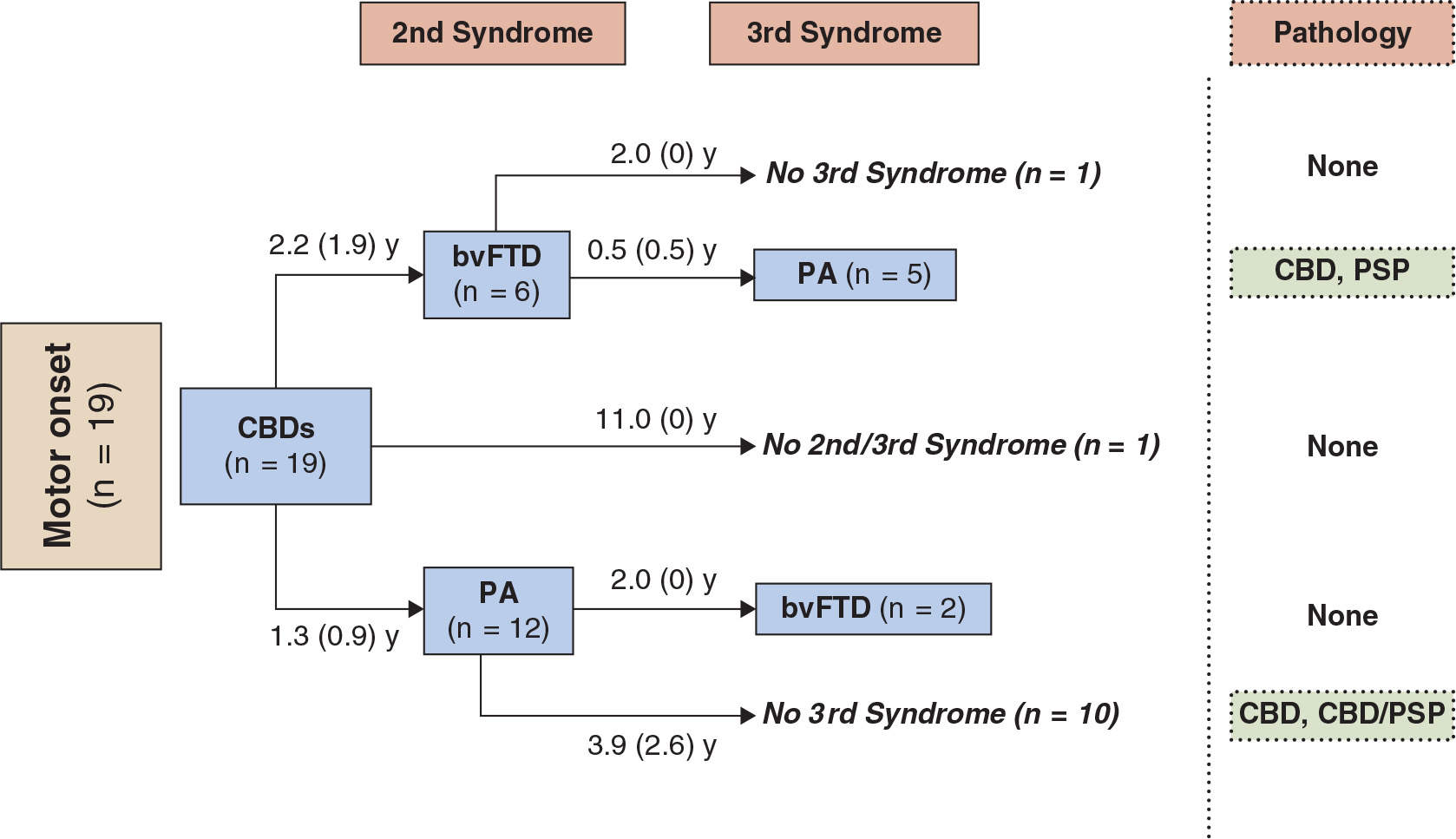
Figure 17.1. The first syndrome and subsequent evolution through second and third syndromes in patients from our center with an initial motor presentation of CBDs. Final pathology is shown where available. Patients developed symptoms meeting criteria for the behavioral variant of FTD (bvFTD) and progressive aphasia (PA). The average interval in years (SD) between the syndromes is indicated. Pathologies are as follows: CBD, corticobasal degeneration; PSP, progressive supranuclear palsy; CBD/PSP, features of both. (From McMonagle P, Kertesz A. Cognition in corticobasal degeneration and progressive supranuclear palsy. In: Miller BL, Boeve BF, eds. The behavioural neurology of Dementia. Cambridge University press, 2009:288–302.)
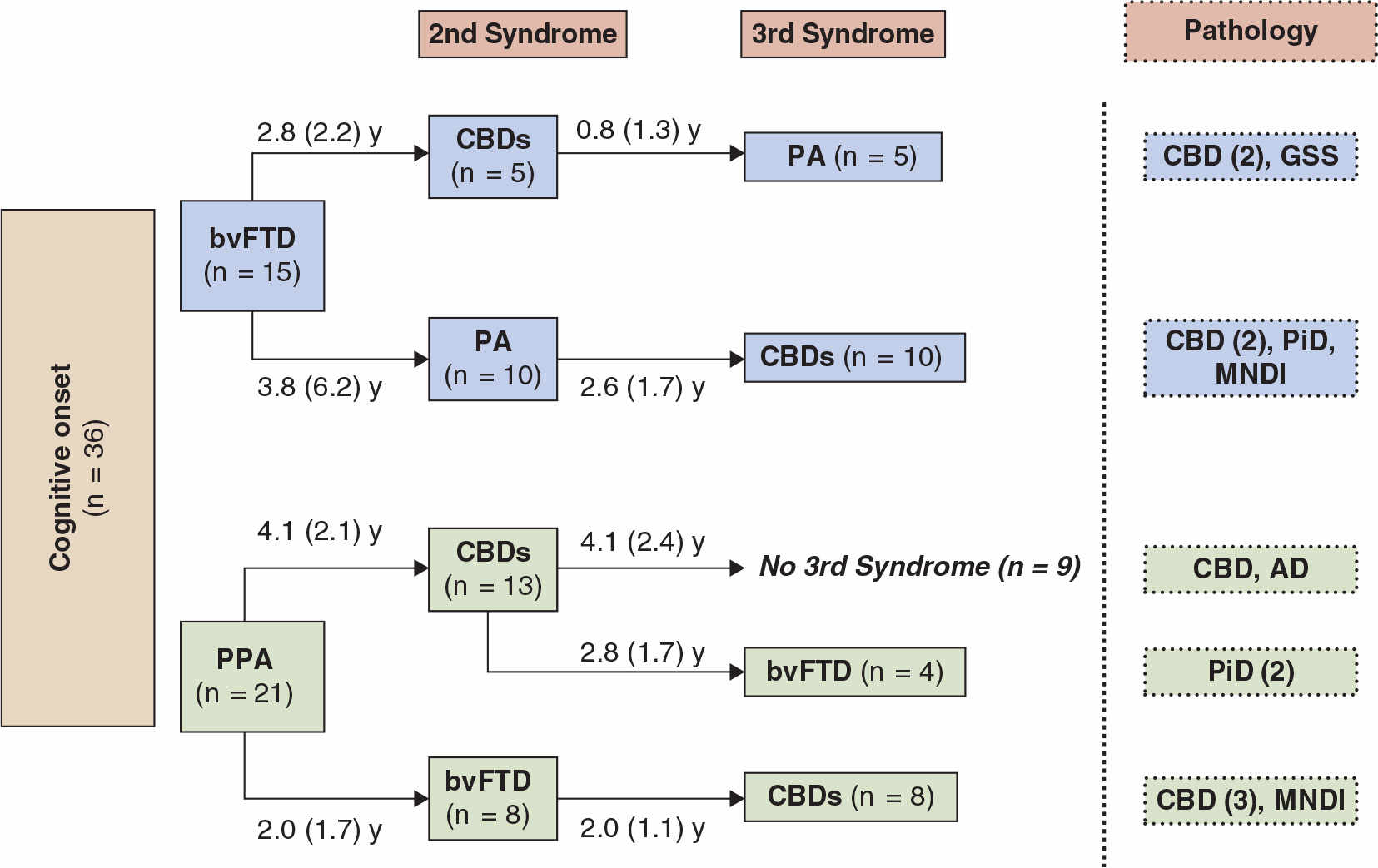
Figure 17.2. The first syndrome and subsequent evolution through second and third syndromes in patients who developed CBDs after an initial presentation with bvFTD or PPA. Final pathology is shown where available. Aphasia developing secondarily is indicated as PA. The average interval in years (SD) between the syndromes is indicated. Pathologies are as follows: CBD, corticobasal degeneration; GSS, Gerstmann–Straussler–Scheinker; PiD, Pick’s disease; AD, Alzheimer’s disease; MNDI, FTD with motor neuron disease type inclusions. (From McMonagle P, Kertesz A. Cognition in corticobasal degeneration and progressive supranuclear palsy. In: Miller BL, Boeve BF, eds. The behavioural neurology of Dementia. Cambridge University press, 2009:288–302.)
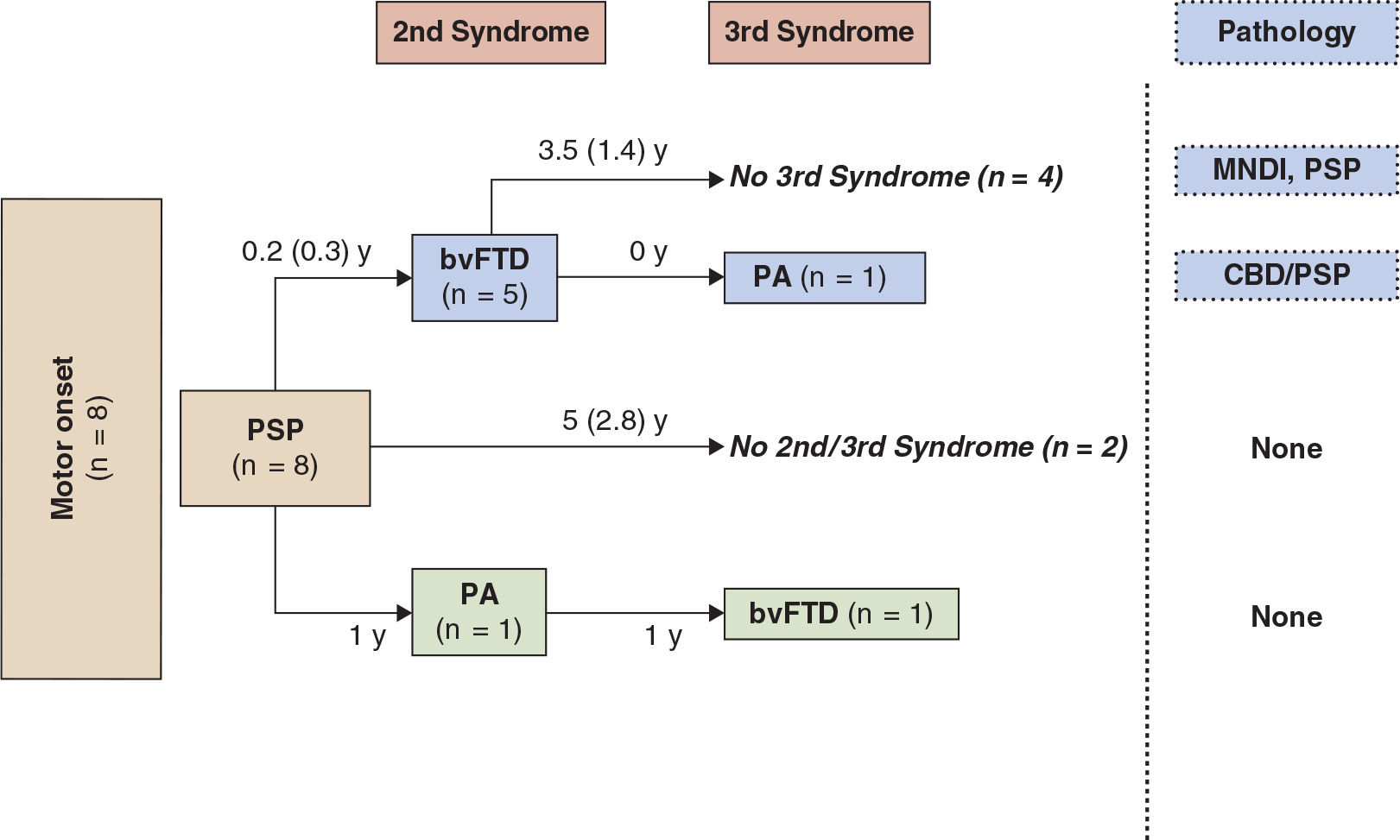
Figure 17.3. The first syndrome and subsequent evolution through second and third syndromes in patients with an initial motor presentation of PSP.
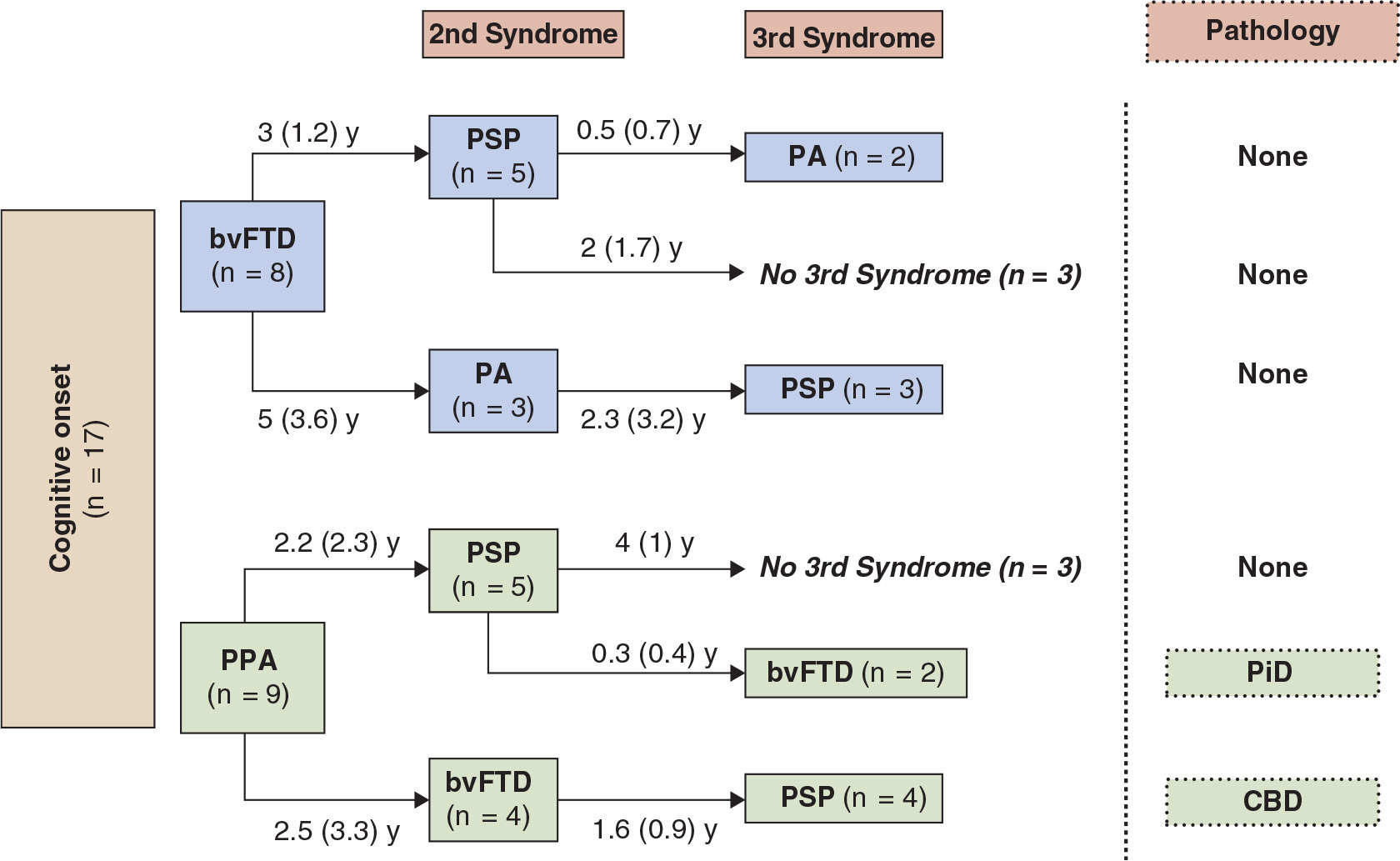
Figure 17.4. First and subsequent syndromes in patients with bvFTD or PPA onsets prior to developing the movement disorder of PSP.
GENETICS
Wilhelmsen et al. (50) discovered linkage to chromosome 17 q21-22 in a large family with variable symptomatology of FTD, aphasia, parkinsonism, and amyotrophy. A consensus conference summarized the clinical and pathologic features of 12 families linked to chromosome 17, and the term “frontotemporal dementia with parkinsonism linked to chromosome 17” (FTDP-17) was accepted (51). The microtubular-associated protein tau (MAPT) gene was suspected as the candidate for mutations, and later several tau mutations were discovered (34,52). The exon 10 splice mutations alter the ratio of 4R to 3R tau isoforms, most often resulting in pathology resembling CBD or PSP. Mutations in exons 9, 12, and 13 result in either accumulation of all six isoforms of tau-forming tangles or in a predominance of 3R tau and Pick body dementia or 4R tau in CBD or PSP. Often, the same mutation such as the common P301L may produce either pathology with Pick bodies or CBD (53). The missense mutations disrupt the interaction between tau and microtubules; unbound tau becomes abnormally phosphorylated and polymerized into filaments and inclusions. Mutations in the tau gene on chromosome 17 can cause genetic forms of CBD with the apparent paradox of the same tau mutation causing bvFTD in the father and CBDs in the son (54). Familial CBDs phenotypes are recognized to overlap chromosome 17-FTLD. The frequency of MAPT was studied in a large cohort of autopsy-confirmed CBD patients (n = 109). Some of these cases with mutation are neuropathologically indistinguishable from sporadic CBD (55). Tau polymorphisms, from the two main haplotypes of tau, indicated that the H1 haplotype is overrepresented in PSP, CBD, and FTD (56).
Progranulin (PRGN) mutation and TDP-43 pathology have been described in CBD/PSP with increasing frequency (32,39,40,42,57–60). PRGN mutations tend to have a parietal syndrome rather than a movement disorder phenotype in CBDs. Familial cases due to mutations in the progranulin gene show a phenotypic overlap, and the same mutation can result in CBDs or bvFTD (58). In some families, PRGN mutations present as behavioral or language syndromes that later progress to a CBDs (60). To further complicate matters, LBD and LRRK mutations are also seen with CBDs/PSPs (61,62).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


