Examples of speech
“We are looking for one hor….like we…are just saying…I should not have done…I didn’t realize it, like I say………. I used to be very ….. I had just first thing I do….here is what she says is …I am giving her problems……. It is me making something I don’t think about.”
Till a few years ago…… I had business…. for saying… because things going wrong because I was dealing people in the house……… I could not get the word. ………Just had to stop.
Examples from written description of the Cookie-Theft Picture test
Boy trying to get cookies. Boy folling of chair.
Mother wiping plate.
Water overflow.
Mother not taken much notice about water on floor.
Cook. Jar
Dishis
Flood Sink
Di
Stool
Anomia is common due to difficulties in word retrieval but object knowledge remains intact. Thus although the patient cannot name an object, they may be able to pick the correct name from choice and will be able to match the object with other objects that semantically linked to it.
Comprehension for single words in usually intact but impaired comprehension of syntactically complex sentences is often an early feature. In addition, these patients have difficulties in speech production due to speech apraxia leading to word sound distortions.
The semantic variant (svPPA, previously semantic dementia) is the second FTD language variant. The hallmark of svPPA is loss of semantic knowledge. This manifests as severe naming and word comprehension impairment in the context of fluent, effortless, and grammatically correct speech output. Semantic paraphasias are common. Words are often substituted by a semantically related but more frequent object (e.g. “horse” for “zebra”) or a more general categorical term (e.g. “animal” for “cow”).
Due to the loss of semantic knowledge, patients have difficulties in single word comprehension and are unable to match the object with semantically similar objects or pick an object from description of its use. Knowledge loss affects initially the less frequent or atypical exemplars of category (e.g. first “hamster”, then ‘rabbit’, then ‘animal’). Knowledge of more personally relevant objects is more resistant. Of note, episodic memory remains intact and patients often remain oriented, are able to keep appointments, and to learn and recall visuospatial information.
Repetition is not impaired in svPPA and patients can repeat multi-syllable words without difficulty. In contrast writing and reading relies exclusively on phonological (letter by letter) reading/writing as patients have no access to previously learned word knowledge, which includes the correct way to read irregular words (such as “yacht” or “cough”). This leads regularisation errors when reading or writing. This is termed as “surface alexia/dyslexia” and “surface agraphia/dysgraphia” respectively.
The above described svPPA phenotype is usually associated with predominant involvement of the dominant hemisphere. A rarer form of svPPA is seen where the disease process affects mainly the non-dominant hemisphere. The phenotype in this form usually includes agnosia (a failure to recognise objects or people) and more prominent behavioural changes.
The logopenic variant of progressive aphasia (lvPPA) is the third type of primary progressive aphasia syndrome. This variant has been recognised only recently and its exact syndromic classification remains controversial. Unlike the other language variants of PPA, the most common pathology underlying this disorder is that of Alzheimer’s type pathology. However, recent evidence suggests that 23 % of lvPPA patient have FTD pathology on autopsy.
lvPPA is characterised by fluctuating interruptions of fluency due to word finding difficulties, with intact grammar and sound production. There is often impaired repetition and comprehension, particularly for long improbable phrases, an observation in line with the hypotheses that episodic memory impairment plays a central role in this condition.
Finally, it is worth noting that while the most salient complaints in language variants of FTD are by definition language related, there are often associated behavioural changes, particularly in svPPA patients. The pattern of behavioural changes in language variant FTD may be different from that of bvFTD. Some reports suggest that apathy, lack of empathy and hyper-orality are more common in bvFTD while compulsive and complex stereotypic behaviours, mental rigidity anxiety, repetitive themes, and sleep disorders may be more common in svPPA.
7.5 Associated Syndromes
7.5.1 Syndromes That Overlap and Mimic FTD
Research has shown the significant pathological and molecular overlap between FTD and other neurodegenerative disorders and hence the crossover in clinical features. Table 7.2 summarizes the syndrome that overlap.
Table 7.2
Clinical syndromes known to display clinical overlap with FTD
Amyotrophic Lateral Scleroses (ALS) | The association between ALS and dementia has been described for over a century. It is now recognised that 13–15 % of FTD patients have evidence of co-morbid ALS (FTD-ALS). Up to 50 % of ALS may have cognitive or behavioural abnormalities on neuropsychological testing which in about 13 % are severe enough to fulfil the criteria for FTD, most frequently bvFTD. |
Corticobasal syndrome (CBS) | This is a progressive disorder characterized by asymmetrical motor and sensory cortical and extrapyramidal dysfunction. It is classically associated with tau pathology though recent evidence suggests that AD pathology underlie many cases. There are two main clinical presentations. In the first one, the patient presents to movement disorders clinic with a “useless arm” due to unilateral rigidity, bradykinesia, apraxia, tremor, and dystonia. In the second, cognitive symptoms, in particular nonfluent progressive aphasia, are the presenting feature. |
Progressive supranuclear palsy (PSP): | This is a rapidly progressing disorders characterized by supranuclear gaze palsy, an akinetic rigid syndrome with pronounced axial rigidity, postural instability causing falls, (mainly backwards), and bulbar symptoms (dysarthria and dysphasia). It is classically associated with PSP pathology. |
On the other hand, there are a range of clinical disorders that have no pathological overlap but whose clinical phenotype can mimic those of FTD. Table 7.3 summarises the syndrome that mimic FTD.
Table 7.3
This is a summary of conditions that can mimic FTD
FTD-Phenocopy Syndrome | There are recent reports of patients who have a typical clinical presentation of FTD (sometimes associated with executive dysfunction and impaired activities of daily living) but they display extremely slow or no progression over time and have normal imaging. The underlying pathology in these patients is yet to be established. It has been suggested that such cases might constitute pathological but stable personality variants |
Psychiatric illness: | Behavioural changes can be interpreted by family and medical professionals as a psychological reaction or psychiatric disorder. For example, apathy may be interpreted as depression and irritability or disinhibition as a midlife personality change. In addition, the negative symptoms characterizing some types of schizophrenia (such as schizophrenia simplex) can be very similar to the behavioural changes of bvFTD, particularly if they are not associated with florid “positive symptomatology”. On the other hand, positive psychotic symptoms such as hallucinations and delusions, though rare have been described in C9orf72 related FTD. Association with other symptoms of FTD and the time course of “insidious onset and gradual progression” can help differentiate FTD |
Alzheimer’s disease (AD) | AD is characterized by early and prominent deficit in episodic memory, and is thus easy to differentiate from FTD. However, atypical variants of AD can be characterized by more prominent involvement of behaviour and language (the so called frontal and language variants) and therefore can pose diagnostic challenges. On the other hand, FTD can also be associated with prominent amnestic features. Notably, attention, orientation, visuospatial skills tend to be preserved in early FTD and impaired in AD. |
Other neurodegenerative disorders | FTD symptoms such apathy irritability and stereotypic behaviour can also be associated with other neurodegenerative disorders affecting the fronto-striatal circuits such as Parkinson’s disease (PD), Huntington’s disease (HD), Wilson’s disease, and Multiple system atrophy (MSA). Olivopontocerebellar atrophy (OPCA) can also present with a range of symptoms reminiscent of FTD. These disorders should have distinguishable features with differing collections of symptoms e.g. tremor in PD, cerebellar ataxia and autonomic failure in MSA and chorea in HD. |
Vascular dementia (VD) | Apathy, executive dysfunction, frontal release signs, and parkinsonian features can occur in VD. However, the stepwise time course and presence of cortico-spinal tract signs would suggest VD over FTD. MRI may be helpful, indicating FTD if there is focal lobar atrophy and VD if there is diffuse white matter changes |
7.6 Clinical Assessment
The first part of the clinical assessment in FTD patients is obtaining a detailed history, emphasising any changes in behaviour, language and other cognitive domains and their impact on the patient’s ability to function at home and at work and to maintain inter-personal relationships. The history should also include a detailed family history and clues suggesting overlap or mimic syndromes (see Table 7.2 and Fig. 7.1). The examination should cover both cognitive functions and motor signs.
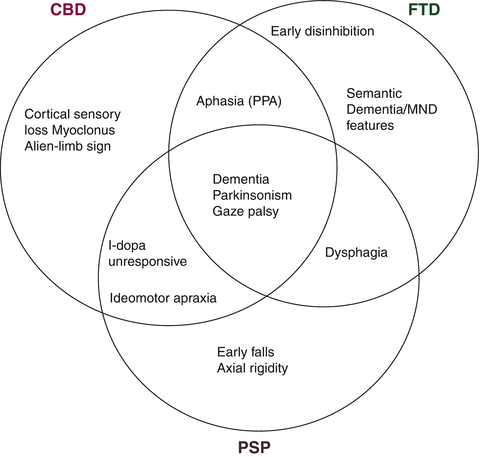
Fig. 7.1
Clinical crossover of frontotemporal dementia, corticobasal degeneration, and progressive supranuclear palsy (Courtesy of Dr. B. Murray, Hermitage Hospital, Dublin)
Cognitive assessment should focus on identifying the cognitive deficits specific to each syndrome in addition to noting which cognitive domains are spared. The most affected domains are executive functions and (in the language variants) language function. In contrast, orientation, calculation, visuospatial skills should be relatively well preserved early in FTD. Although early memory impairment is generally considered to exclude a diagnoses of FTD, recent evidence suggest that memory changes in FTD are common, especially in younger patients. Occasionally, memory dysfunction occasionally can be as severe as that observed in AD patients. In fact, severe amnesia can be the presenting feature in some patients. However, typically memory function in FTD affects free recall to a higher degree than recognition or cued recall while in AD the impairment extends to all three aspects of episodic memory.
A variety of rapid screening tests have been designed specifically for the purpose of discriminating FTD from non-FTD dementias such as the third version Addenbrooke’s Cognitive Examination (ACE-III, Hsieh 2013) which is the most updated version of the ACE (Mathuranath 2000), the Executive Interview (Royall, Mahurin RK, and Gray, 1992), and the Frontal Assessment Battery (FAB, Dubois, Slachevsky, Litvan and Pillon B, 2000), and The Montreal Cognitive Assessment (MoCA, Nasreddine et al. 2005). If feasible, the patient can be referred to a clinical psychologist for a more detailed neuropsychological evaluation.
The motor examination should focus on the presence of motor signs suggesting disease mimics (e.g. stroke) and disorders that overlap with FTD such as corticobasal syndrome (CBS), Progressive Supranuclear Palsy (PSP), and amyotrophic form of Motor Neuron Disease (FTD-ALS). Thus presence of extra-pyramidal signs, muscle wasting, fasciculations or unilateral apraxia add to the behavioural and cognitive profile in characterizing the syndrome (Fig. 7.1).
7.7 Neuropsychological Assessment
Given the wide variability of presentations in FTD and the fact that there are a number of other overlapping conditions, a comprehensive neuropsychological assessment can greatly contribute to differential diagnosis. However, as FTD can frequently present with alterations in behaviour and personality one should not over-rely on cognitive testing per se. Depending on the locus of pathology patients may do extremely poorly on conventional “frontal lobe” tests (such as the Wisconsin Card Sorting Test, Trail Making Test, verbal fluency etc.), or they may in fact perform normally, yet still show major impairment in self- and social regulation. Therefore it is essential to conduct a detailed evaluation of behaviour.
Behavioural evaluation should include direct observation of the patient as well as collateral information from informants using semi-structured interviews and preferably a suitably designed behavioural instrument. One such instrument is the Frontal Systems Behaviour Scale (FrSBe, Grace & Molloy 2001) which was designed based on Cummings’ neuroanatomical model to provide self and informant based ratings of apathy, disinhibition, and executive dysfunction both pre-morbidly and currently. In addition to permitting age and gender graded interpretation of behaviour change, the self- and informant-based ratings can be compared to evaluate the degree of insight or lack thereof. Other commonly used behavioural instruments include the Cambridge Behavioural Inventory (CBI, Wedderburn et al. 2008) and the Neuropsychiatric Inventory Questionnaire (NPI-Q, Kaufer et al. 2000).
The typical pattern of cognitive change reported in FTD patients is impairment of executive functions. There are purpose designed comprehensive batteries to evaluate a broad range of cognitive processes encompassed within the term executive function, the majority of which are dependent on the integrity of the frontal lobes. Notable examples include the Delis-Kaplan Executive Function System (D-KEFS, Delis, Kaplan, & Kramer, 2001) and Behavioural Assessment of the Dysexecutive Syndrome (BADS, Wilson, Alderman, Burgess, Emslie, & Evans, 1996), (see Chap. 3 for a more detailed discussion of tasks of executive functions).
As noted above, the patient may or may not have associated significant memory impairment. Attention, orientation and visuo-spatial functions are typically preserved in FTD patients. However, it is worth noting that qualitative cognitive analysis is as important as the patient’s quantitative performance on the different neuropsychological tests. Patients can fail the same tests for different reasons. For example, an AD patient could fail visuo-construction tasks such as copy trial of the Rey Osterrieth Complex Figure or Block Design because of memory problems and/or poor visuo-spatial functions while FTD patients might have difficulties with the same task due to constructional praxis and planning problems.
Patients with an early disease focused in the orbitofrontal-ventromedial prefrontal cortex, may perform normally on tests traditionally considered sensitive to frontal lobe/executive dysfunction but have a profound deficit in everyday decision making and social regulation. To address this issue novel batteries have been developed to focus on emotional decision making and social cognitive skills. Many tasks intend to mimic every day functioning and as such maybe more sensitive to cognitive changes in early bv-FTD.
One such battery is the Executive and Social Cognitive Battery (ESCB, Torralva 2009) which includes the Iowa Gambling Task (IGT, Bechara et al. 1994), the Multiple Errands Task (MET, Shallice & Burgess, 1991), the Hotel task (HOT, Manly et al. 2002), the Mind in the Eyes task (Baron-Cohen et al. 2001), and the Faux Pas test (Stone et al. 1998). The IGT evaluates decision making and learning in high and low risk situations. During the task healthy individuals learn to avoid the risky choices while those with FTD continue to make high risk choices which results in an overall net loss. The MET and the HOT are both designed to mirror everyday tasks. The MET involves the participant running “real life” errands such as purchasing three items (a soda, a postcard and a letter), using the internal phone, and obtaining and writing down pieces of information such as the area code of a city. The Hotel task involves activities that are often undertaken as part of running a hotel such as sorting bills by client name. The Mind in the Eyes task requires the participant to choose from four options the word that most accurately describes how a set of individuals are feeling based on photographs of their eye region. The Faux Pas test is based on “theory of mind” (the ability to infer another’s thoughts and feelings based on social cues). Changes in this ability may underlie some of the changes in personality and social functioning frequently seen in FTD patients. The task entails hearing 20 short stories, 10 of which contain a social faux pas and 10 of which are neutral. Following each story, questions are asked to evaluate the patient’s social awareness and understanding. Patients with bvFTD, but not patients with AD do poorly on this test.
7.8 Neuropathology
The neuropathology associated with the clinical entities of FTD is heterogeneous with the unifying feature being a relatively selective and impressive atrophy of the frontal and/or temporal lobes on macroscopic examination of the brain. The pattern of atrophy in FTD is linked to the clinical phenotype: mainly left hemispheric atrophy in nfvPPA, bilateral (often asymmetrical) atrophy of the middle and inferior temporal lobes in svPPA, while in bv-FTD there is usually early bilateral orbito-mesial atrophy, followed by atrophy of the dorsolateral frontal cortex, temporal pole, hippocampal formation, and the basal ganglia.
Importantly, the lack of macroscopic atrophy does not exclude microscopic evidence of FTD pathology. Crucial also is the observation that microscopic neuropathological changes in FTD do not map onto clinical features in a one-to-one manner. The association of a highly specific constellation of symptoms and signs with more than one neuropathological signature may seem paradoxical but it may be understood in terms of systems neurodegeneration i.e. degeneration of neuronal populations that are connected structurally and functionally.
The microscopic appearance of the cortex in FTD patients includes neuronal loss, widespread spongiosis and astrocytosis obscuring normal pathology. In addition, as in other neurodegenerative conditions, FTD is characterised by specific kinds of intracellular protein inclusions. In 1911 Alois Alzheimer described round silver-impregnated inclusions and swollen neuronal perikarya (cell bodies) in cases of dementia with prominent language and behavioural symptoms, first described clinically by Arnold Pick in 1892. The inclusions would become known as “Pick Bodies”, the defining histopathological lesion of Pick’s disease.
In the last 30 years there has been significant progress in the neuropathology of FTD. This prompted the Midwest Consortium for Frontotemporal (Lobar) Degeneration and other groups to review the existing neuropathological diagnostic criteria for FTD. Currently, the heterogeneous group of disorders referred to under the umbrella term of FTD are categorized pathologically based on the biochemical composition of the observed protein inclusions.
7.8.1 Tau
Tau is a phosphorylated protein expressed predominantly in neurons. The phosphorylation of tau is believed to be critical to its ability to bind and stabilize neuronal microtubules which are cytoskeletal structures of axonal transport.
Tauopathies are characterized by deposits of tau protein and represent approximately 40–50 % of sporadic FTD cases. A tauopathy is almost always observed in post mortem studies of patients with nfvPPA, FTD with Pick bodies (Pick’s disease), and patients with BVFTD and parkinsonism linked to mutations in the microtubule-associated protein tau (MAPT) on chromosome 17 (FTDP17, see Sect. 7.9 below). Behavioural or language variant FTD patients who exhibit extrapyramidal signs often have tau pathology. Other tauopathies include cortico-basal degeneration (CBD), progressive supranuclear palsy (PSP), argyrophilic grain disease, and neurofibrillary tangle dementia (Tangle-only dementia). Although tau pathology is also present in AD, this disease is usually not referred to “tauopathy” since it always associated with co-existing amyloid pathology.
Pick bodies as originally described by Alzheimer are now known to be spherical cytoplasmic inclusions that are tau positive. Pick bodies are typically found in the cingulated gyrus, insula, inferior parietal lobule and inferior temporal gyri. They are also found in the mesial structures particularly the granule cells of the dentate fascia. White matter pick bodies are more common in CBD and PSP but can be found in FTD. Pick bodies may also be found in the basal ganglia and substantia nigra.
7.8.2 TDP-43
Many FTD cases are tau negative. Until recently, ubiquitin immunohistochemistry was the only method to detect the abnormal protein inclusions in the vast majority of these cases, prompting the term frontotemporal dementias with ubiquitin inclusions (FTLD-U), which is thought to represent about 60 % of FTD cases.
A major discovery in 2006 by Neumann and colleagues revealed that the main constituent of ubiquitin inclusions in up to 90 % of FTLD-U cases is a protein called trans-activating responsive DNA-binding protein 43 (TDP-43). It is now recognized that almost all svPPA and FTD-ALS patients as well as 50–60 % of bvFTD patients are TDP-43 positive. Of note, the vast majority of patients with sporadic amyotrophic lateral scleroses (ALS) and some familial ALS patients are also positive for TDP-43 pathology. This discovery was a major milestone in understanding the degree of overlap between ALS and FTD and led to the emergence of a new family of neurodegenerative disorders, the so called TDP-43 proteinopathies, which includes ALS as well as FTLD-U with and without ALS.
TDP-43 is a ubiquitously expressed protein encoded by the TARDBP gene on chromosome one. It is a nuclear protein, but it shuttles between the nucleus and the cytoplasm with normally low concentrations in the cytoplasm. Both the structure and cellular distribution of TDP-43 is abnormal in TDP-43 proteinopathies. TDP-43 loses its nuclear position and aggregates mainly in cytoplasm. The TDP-43 protein itself is truncated at the N-terminus while the remaining C-terminus fragment is abnormally phosphorylated to form insoluble cytoplasmic protein deposits observed in the brain and spinal cord in FTLD-U and sporadic ALS.
Brains with TDP-43 pathology can display neuronal cytoplasmic inclusions (NCIs), neuronal nuclear inclusions (NNIs) and/or dystrophic neurites (DNs). These changes have been described in the neocortex and hippocampus and the lower motor neurons in both FTLD-U and ALS. On other hand, TDP-43 positive glial inclusions (GI) were mainly considered a feature of ALS, but have been described in FTLD-U.
There is pathological heterogeneity within the TDP-43 proteinopathy family with more than one classification proposed. The harmonized classification system, recently published by Mackenzie et al. in 2011, described four main subtypes of TDP-43 pathology (see Table 7.4).
Table 7.4
This table provides a summary of the harmonised classification (Mackenzie 2011) of pathological subtypes within the family of TDP-43 proteinopathies
Subtype Mackenzie 2011 | Characteristic Pathology | Previous Nomenclature | ||
|---|---|---|---|---|
Inclusions/Cellular changes | Cortical distribution | Mackenzie | Sampathu | |
Sub-type A | NCIs Abundant NCIs ± Moderate numbers of lentiform NIIs Many crescentic or oval DNs | Mainly layer II of cortex | Type 1 | Type 3 |
Sub-type B | Moderate NCIs Occasional NIs DNs rare | Affects all cortical layers | Type 3 | Type 2 |
Sub-type C | Very Few NCIs Abundant long DNs | Mainly layer II of cortex | Type 2 | Type 1 |
Sub-type D | Rare NCIs, Abundant lentiform NIIs Abundant short DNs | Affects all cortical layers | Type 4 | Type 4 |
The pathological subtypes summarized in Table 7.4 have distinct clinical and genetic correlations. In subtype A, the clinical presentation is usually bvFTD or nfvPPA (rarely svPPA) with high prevalence (up to 50 %) of familial disease. This subtype is consistently reported in patients with GRN gene mutations and has recently been reported in C9orf72 gene mutations.
Subtype B is associated a clinical picture of FTD-ALS or bvFTD and is associated with generally poor prognoses. Genetically, subtype B is linked to mutations in the C9orf72 gene.
Subtype C is associated with the svPPA variant of FTD and less commonly bvFTD but there are no genetic associations to date.
Subtype D is associated with familial Inclusion body myopathy with Paget Disease of Bone and frontotemporal dementia (IBMPFD) which is due to mutations in the valosin-containing protein (VCP) gene.
Detailed discussions of these gene mutations are included in the Sect. 7.9 below.
Of note, the authors of the new classification highlighted its limitations which include failure to incorporate sub-cortical changes or describe the TDP-43 pathology recently reported in normal aging population, in the Guam Parkinson Dementia Complex patients, in about 20 % of AD patients, and up to 70 % of patients with hippocampal scleroses with and without AD as demonstrated by Amador-Ortiz et al. (2007).
7.8.3 FUS
Up to 10 % of FTLD-U patients are not only tau negative but also TDP43 negative. In addition, 15 % of ALS-FTD cases do not belong to the FTLD-U subtype and do not stain with ubiquitin or TDP-43. Interestingly, most cases of familial ALS associated with mutations in the superoxide mutase gene are also TDP-43 negative.
In 2009, the inclusions in some TDP-43 negative FTLD-U cases and some familial ALS cases are found to be positive for Fused in Sarcoma (FUS) protein. FTLD-U patients who are positive for FUS protein are termed FTD-FUS.
FUS is a protein involved in RNA processing and is strikingly similar to TDP43 in in structure and function. Nuclear FUS is believed to play an essential role in regulation of transcription and pre-mRNA splicing while in the cytoplasmic FUS is likely to be involved mRNA transport.
All reported cases of FTLD-FUS have been associated with a clinical diagnosis of bvFTD (with or without the signs of MND).
7.8.4 Rare Subtypes
A small number of tau negative FTLD-U cases are negative for TDP-43 and FUS. These are referred to as FTLD- ubiquitin proteasome system (FTLD-UPS).
Other rare types of FTD pathology include basophilic inclusion body and neuronal intermediate filament inclusion disease.
There are also FTD cases that are negative for tau as well as ubiquitin. They form a rare and controversial entity referred to a Dementia lacking distinctive histopathology (DLDH). This previously large group of identified pathology has been gradually replaced by newly identified pathological variants (Fig. 7.2).
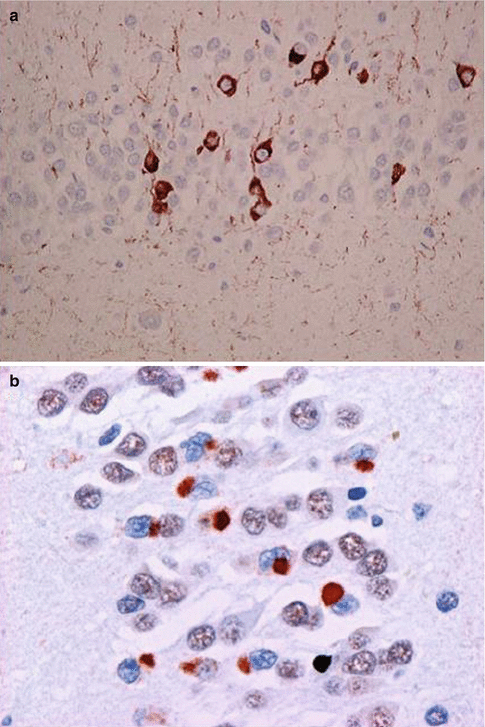
Fig. 7.2
Microscopic staining in FTD. (a) Tau-positive staining in FTD17-T (arrow) (Courtesy Prof. Michael Farrell); (b) TDP-43 staining in FTD-U/ALS (arrow) (Courtesy Prof. Ian R. A. Mackenzie)
7.8.5 Clinical Phenotype and Neuropathology
As noted in the beginning of this section, the clinical presentation in FTD does not map in a one-to-one manner with the clinical phenotypes. The exceptions to this general rule are FTD-ALS which is generally positive for TDP-43. In a recent study by Chare and colleagues (2014) the newly proposed clinical classifications of FTD were retrospectively applied to 135 autopsy ascertained FTD cases from the Cambridge Brain Bank (UK) and the Sydney Brain Bank (Australia). The brain pathology underlying different FTD syndromes in that cohort was as follows:
bvFTD patients :42 % FTLD-Tauopathy, 30 % FTLD-TDP-43 proteinopathy, 13 % FTLD-FUS, 9 % Alzheimer’s type pathology, and 6 % other rare FTLD pathologies;
svFTD : 68 % FTLD-TDP-43 proteinopathy while both FTLD-tauopathy and AD type pathology were responsible for 16 % of cases each;
nfvPPA: 50 % FTLD-tauopathy, 31 % AD type pathology, and 19 % TDP43-proteinopathy; and
lvPPA: 77 % AD type pathology, 14 % TDP-43 proteinopathy and 9 % FTLD-tauopathy.
7.9 Genetics
The progress in this area, moribund for decades despite the recognition of the important of heritability since the 1920s, has been rapid and ever-expanding in the last two decades. The traditional disease dichotomy of ‘sporadic’ versus ‘genetic’ is still used but is increasingly difficult to support, due to the likely polygenic factors influencing sporadic FTD. Nevertheless, the first step in determining whether there is a genetic influence in a disorder is to establish the frequency of a family history of the disorder.
The earliest well documented large pedigree was first reported in 1939 by Sanders and Colleagues who described an autosomal dominant dementing disorder with behavioural and cognitive disturbances with relatively preserved memory affecting a Dutch Kindred. However, the earliest estimates of the frequency of family history came from the Lund and Manchester clinico-pathological series, which estimated that up to 50 % of patients with FTD had a first degree relative with dementia. More recent studies have confirmed that about 30–50 % of FTD patients have family histories of dementia. However, the accuracy of ascertainment of familial disease is confounded by informant reliability, the late onset of the disease, the possibility of death before disease expression, and the fact that informants often report a vague history of “dementia” of unknown type in elderly relatives. It is also worth noting that the variable phenotype of FTD seems to have a direct effect of reported rates of familial disease with highest rates reported in FTD with co-morbid ALS (up to 60 %) and lowest rates reported in svPPA variants of FTD (20 % or less).
The main genes are associated with familial FTD are listed here in chronological order of discovery
1.
MAPT gene on chromosome 17, (1998),
2.
VCP gene on chromosome 9, (2004),
3.
CHMP2B gene on chromosome 3, (2005),
4.
GRN (or PGRN) gene on chromosome 17 (2006),
5.
TARDBP gene on chromosome 1, (2008),
6.




C9orf72 gene on chromosome 17, (2011),
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
