Advances in imaging have improved our understanding of several neurologic disorders; this is perhaps especially true for idiopathic Parkinson’s disease (PD), and other disorders of basal ganglia function, where structural changes are often quite limited. Functional imaging involves the use of radionuclide imaging techniques, such as positron emission tomography (PET) and single photon emission computed tomography (SPECT), or functional magnetic resonance imaging (fMRI). These powerful techniques have enabled the in vivo study of neurotransmitter and receptor function, alterations in blood flow and metabolic activity, functional connectivity, abnormal protein deposition, and inflammation, thereby providing valuable insights into nervous system physiology, and its alteration in disease. Complementary to functional imaging is the rapidly growing capacity to image brain structure using novel MRI techniques. These developments have led imaging to become the strongest current potential biomarker for various parkinsonian disorders. Although the diagnosis of parkinsonian disorders continues to be clinical, the role played by imaging is expanding. In this chapter, we review the literature on functional imaging in movement disorders, with an emphasis on radionuclide imaging techniques, especially PET. Some of the studies performed using PET can also be done with SPECT.
PRINCIPLES OF PET AND SPECT
In both PET and SPECT, biologic processes are studied using radiopharmaceuticals. In PET, the molecule of interest is labeled with a radioactive isotope that is usually neutron-deficient, rendering it unstable. Stability is achieved by the conversion of a proton into a neutron, with the discharge of a positron (positively charged electron) and a neutrino. The positron travels a short distance, until it encounters an electron, resulting in annihilation and the release of two photons or γ rays, which are emitted at 180°. These can be detected with the help of ring detectors. Photons that arrive coincidentally, at detectors opposite to each other, are deemed to have been released from the same annihilation reaction, and the information can be reconstructed to provide a time-dependent image of retained radioactivity. Interpretation of the radioactivity is dependent upon an understanding of the biologic processes being assessed and application of an appropriate mathematical model, which will vary depending upon the nature of the process itself, as well as the properties of the tracer, including the presence or absence of radiolabeled metabolites, whether or not they cross the blood–brain barrier, and whether or not the tracer binds in a reversible or irreversible fashion during the course of the scan. Some tracers require an arterial line in order to obtain an appropriate input function, whereas for others, the use of a tissue reference region may suffice. Various radioactive isotopes with different half-lives can be used. The most widely used radioisotopes with their approximate half-lives in parentheses are 15O (2 minutes), 11C (20 minutes), and 18F (2 hours).
In contrast to PET, SPECT imaging uses radioisotopes which directly emit single photons or γ rays. These single photons are imaged with the help of a γ-camera which rotates around the subject. This is different from PET, in which detection of coincident events is the basis for image formation, but similar to conventional computed tomography (CT), although the latter is clearly based upon an external source of radiation. To ensure that the camera is detecting photons from a single plane, collimators are used in SPECT. Historically, image resolution in SPECT has been inferior to PET. With newer generation cameras the resolution of SPECT images has improved considerably. The performance of a transmission scan using an external source of radiation allows the accurate measurement of attenuation of radioactivity for each voxel with PET, while SPECT techniques generally depend upon an estimation of this variable. On the other hand, PET radioisotopes have a shorter half-life and therefore require close proximity to a cyclotron, which not only limits the widespread utility of this technique but also raises its cost.
PARKINSON’S DISEASE
DOPAMINERGIC IMAGING
The generation of dopamine from its precursor levodopa (L-dopa) by the enzyme aromatic amino acid decarboxylase (AAAD), its storage into synaptic vesicles by the vesicular monoamine transporter 2 (VMAT2), and its reuptake from the synaptic cleft by the membrane dopamine transporter (DAT) have all been studied using appropriate radio ligands (Fig. 45.1). Imaging these biologic processes in the striatum provides an indirect measure of the integrity of presynaptic nigrostriatal dopamine neurons. In PD, pathologic studies have demonstrated that the loss of nigrostriatal dopamine neurons is generally asymmetrical, and that projections to the posterior striatum are more affected than the anterior. Functional imaging has demonstrated a similar pattern in vivo, irrespective of the tracer and biologic process being evaluated (see Fig. 45.1). However, although results are comparable, there are subtle differences between tracers, due in part to compensatory changes which arise when these processes are affected. 18F-fluoro-L-dopa (18F-FDOPA) is converted by AAAD into 18F-fluorodopamine and stored in synaptic vesicles. Its uptake in PD is higher than expected, when compared to VMAT2 binding, which is studied with 11C- or 18F-labeled dihydrotetrabenazine (DTBZ). This is postulated to result from a compensatory increase in the activity of AAAD (1). In contrast, DAT binding, which generally has been studied with either cocaine-like derivatives or methylphenidate, is lower than expected, when compared to VMAT2 binding. This is possibly due to downregulation in the number of DAT, to ensure higher concentrations of dopamine in the synaptic cleft (1). The influence of compensatory mechanisms is thought to be the least for VMAT2 binding (2,3), although theoretically, this too could be influenced by the concentration of dopamine in the presynaptic vesicle (4,5).
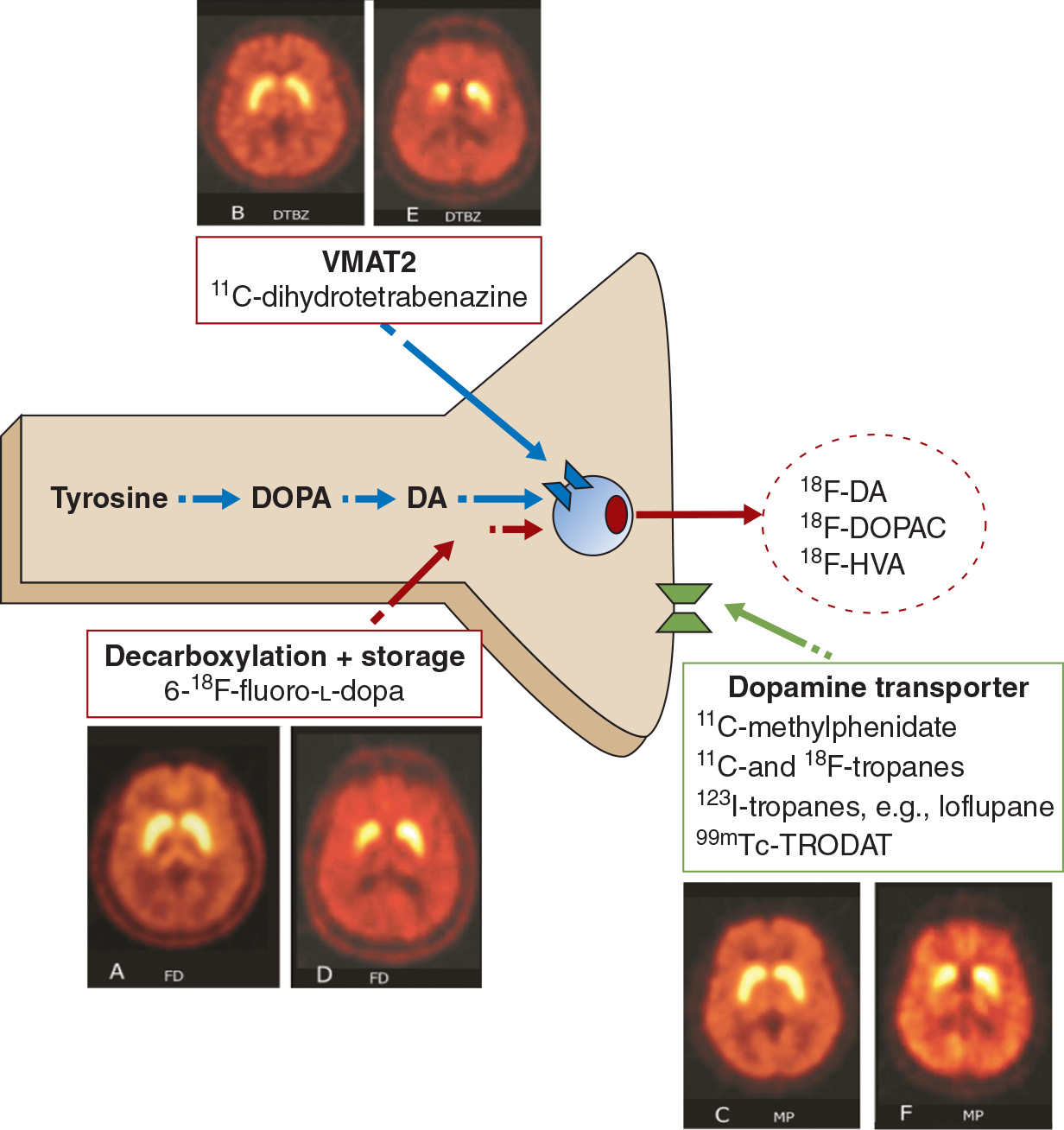
Figure 45.1. Assessment of presynaptic dopaminergic function. The vesicular monoamine transporter (VMAT2) is labeled with 11C-dihydrotetrabenzine or its 18F analog. Decarboxylation and storage of levodopa is assessed with 6-18F-fluoro-L-dopa (FD), and the dopamine transporter is assessed using a variety of 123I-, 99mTc (SPECT)-, 11C-, or 18F (PET)-labeled tropanes or with 11C-d-threo-methylphenidate (MP). For each approach, representative images are shown from a healthy control (left) and a subject with early Parkinson’s (right).
Dopamine receptors, which are classified as either D1-like (D1, D5) or D2-like (D2, D3, D4), can also be imaged using radiotracers with affinity to the receptor subtype of interest. In parkinsonian syndromes, the D2-like receptors have been studied more extensively. The signal measured, termed the “nondisplaceable binding potential” (BPND) (6), is a function of the receptor density and affinity. Hence, a change in either of these parameters will alter the final measurement, and should be taken into consideration when interpreting results of a single study. Theoretically, binding could be affected by internalization of receptors following sustained stimulation; however, this does not appear to be the case in practice (7). Endogenous dopamine competes with low affinity radiotracers, such as 11C-raclopride for receptor binding (8). Dopamine release can be estimated by measuring radiotracer binding in serial studies, before and after exposure to the stimulus of interest. If the stimulus were to result in release of dopamine, the binding of radiotracer would decrease, because of competition from endogenous dopamine. In contrast, radiotracers with high affinity to the D2 family of receptors are utilized to image dopamine receptors in extrastriatal regions where they are found in low concentrations. In early untreated PD, increased binding to D2-like receptors has been shown, and this probably results from a combination of receptor upregulation and reduced competition from low levels of endogenous dopamine. In contrast, in atypical parkinsonian disorders, the binding to D2 and D1 receptors is decreased (9–11), and this probably results from a loss of postsynaptic striatal neurons. In PD, following treatment, D2-receptor binding returns to normal or slightly less than normal (12). D1-receptor binding is unaltered in PD, but these receptors may have a role in the development of dyskinesia.
IMAGING OF OTHER NEUROTRANSMITTERS
Although dopamine is the major neurotransmitter involved in PD, abnormalities in other neurotransmitter systems have been found. 18F-FDOPA is also taken up by monoaminergic neurons in the locus coeruleus (LC), which is predominantly noradrenergic, and in the median raphe nucleus, which is predominantly serotonergic, thereby providing a measure of activity in these nuclei. In early PD, there is increased 18F-FDOPA uptake in the LC, which probably reflects compensatory upregulation of AAAD; uptake declines with progression of illness (13). In addition, the uptake of RTI-32, an agent which binds to both the norepinephrine transporter and the DAT, is reduced in several brain regions such as the LC and thalamus in PD with depression and anxiety, in comparison to PD patients without these comorbidities. The changes in the LC and thalamus are most likely related to reductions in the activity of norepinephrine transporter (14), although there are dopaminergic projections to thalamus.
Serotonin transporter activity has been studied in PD using 11C-3-amino-4-(2-dimethylaminomethyl-phenylsulfanyl)benzonitrile (11C-DASB), and has been shown to be reduced in the striatum, brain stem, and cerebral cortex. This reduction did not correlate with disease duration, severity, or medication use (15). In rodents, serotonergic neurons in the striatum are postulated to play a role in the development of dyskinesia, by the uptake of L-dopa, and consequent unregulated release of dopamine (16). Increased serotonergic innervation has also been shown to occur in PD patients treated with fetal mesenchymal stem cells and graft-induced “off” dyskinesia (17), and more recently in L-dopa–induced “on” dyskinesia (18). Interestingly, both types of dyskinesia responded to 5-HT1A agonists, by reducing the release of dopamine (17,18). One would expect low levels of serotonin transporter activity in patients with PD who are depressed; however, 11C-DASB has been found to be increased in this subgroup, in comparison to those without depression (19) and even to normal controls (20). The bases of these findings are presently unclear.
Cholinergic activity has been studied using radioligands, such as 11C-1-methylpiperidin-4-yl propionate (11C-PMP) and [11C]-N-methylpiperidin-4-yl acetate (11C-MP4A), which are lipophilic and therefore able to cross the blood–brain barrier, but are hydrolyzed by acetylcholinesterase to hydrophilic metabolites and trapped within the brain (21). Reduced cholinergic activity, assessed using this technique, has been demonstrated in the cerebral cortex of patients with PD, more so in patients with PD dementia. This reduction is in fact greater than what has been shown in Alzheimer’s disease (AD) (21). Moreover, in PD, reduced cholinergic activity in the thalamus has been related to falls (22), whereas reduced cholinergic activity in cortex has been correlated with reduced gait speed (23). Nicotinic acetylcholine receptors (nAchR) have also been studied in PD. One study showed a widespread reduction in nAchR in patients with PD as compared to normal controls, with regional differences in those with depressive and cognitive symptoms (24).
PD patients with dyskinesia have been found to have low levels of opioid receptors (25), and high levels of adenosine A2A receptors in the striatum (26), in comparison to those without dyskinesia. Increased binding of cannabinoid receptors has been shown in the striatum, and the projection areas of the mesolimbic and mesocortical pathways in PD (27).
METABOLIC PATTERNS
Regional cerebral blood flow or glucose metabolism is known to be indicative of local neuronal and synaptic activity. Alterations in such activity may often precede structural changes observed on routine imaging, as is the case in many neurodegenerative disorders. Initial enthusiasm for the study of brain metabolism was tempered by the large amounts of data generated when using these techniques, as well as the great variability within a subject, and between different brain regions, which made analysis complicated. This led to most initial studies using hypothesis-driven approaches, wherein brain regions of interest were compared between subject groups. Over time, data-driven statistical methods have been developed. Broadly, two types of analysis are used. In statistical parametric mapping (SPM), voxel-by-voxel comparisons of metabolic activity are made between patients and controls without a priori hypothesis. However, while useful for detecting unpredicted regional group differences in metabolism or blood flow, this technique has a number of technical and statistical challenges, and is not geared toward the study of systems-level or network abnormalities. This limitation is especially relevant in the context of neurodegenerative disorders, which may be associated with characteristic alterations in connectivity affecting certain brain regions. One technique that enables the study of networks is the scaled subprofile model (SSM), essentially a form of principal components analysis. Using this technique, the spatial covariance pattern of metabolic imaging data can be expressed as a single numerical (28). This is a significant advantage of this technique, and allows for the study of disease progression, and response to treatment (28). Based on spatial covariance analysis of 18F-fluoro-deoxy glucose (18F-FDG) PET images, an abnormal metabolic network has been identified in PD in several studies (28). This is known as the PD-related metabolic pattern (PDRP) (Fig. 45.2). It is characterized by increased metabolic activity in the pallidum, thalamus, and pons, with reductions seen in the premotor cortex and parietal association areas (28). The expression of the PDRP is attenuated by treatment with L-dopa or deep brain stimulation (29) (see Fig. 45.2). In the absence of medication, PDRP expression correlates in both scans of cerebral blood flow and glucose metabolism (30,31). The expression of PDRP is unaltered by the presence or absence of parkinsonian tremor. A PD tremor–related pattern (PDTP) characterized by increased metabolic activity in the cerebellum, dorsal pons, primary motor cortex, and putamen has been described, it is specifically related to the severity of parkinsonian tremor, and unrelated to bradykinesia and rigidity (32). A network related to cognitive dysfunction in PD has also been identified, characterized by reduced metabolic activity in the medial prefrontal, premotor, and parietal association areas, along with increased activity in the cerebellar vermis and dentate nuclei. This metabolic pattern has been termed the PD-related cognitive pattern (PDCP) (33).
IMAGING AS A MARKER OF PRECLINICAL DISEASE AND PROGRESSION
By the time PD manifests clinically, approximately 50% of dopaminergic neurons have already been lost and dopaminergic activity in the striatum is reduced by around 80%. Although these numbers demonstrate the brain’s remarkable reserve, they also emphasize that patients seen for the first time in the clinic may actually have fairly advanced disease from the pathologic and neurochemical perspective. This has important implications for future disease-modifying therapies, as some of these therapies may have potential benefit limited to early disease. Imaging can play an important role in identifying preclinical disease. PET imaging has been used to show dopamine deficits in the clinically unaffected twin of monozygotic twin pairs; the deficit progressed on longitudinal follow-up, with some subjects developing clinically manifest PD (34). Dopaminergic deficits have also been shown in subjects exposed to the nigral toxin MPTP, without manifest parkinsonism (35), and in asymptomatic individuals with a family history of dominantly inherited PD (36). Interestingly, dopaminergic deficit has also been reported in asymptomatic heterozygote mutation carriers of the parkin (PARK2) and PARK6 genes (37,38), which are responsible for recessively inherited PD. The significance of the latter findings is unclear. Subclinical deficits in dopamine function have also been shown in subjects who are asymptomatic for PD, but considered at risk because of hyposmia (39) or rapid eye movement sleep behavior disorder (RBD) (40).
Imaging can play an important role as a biomarker to study progression of PD. In a longitudinal PET study of patients with PD, markers of presynaptic dopaminergic integrity were found to decline, and were best described using an exponential function which approaches a nonzero asymptote (41) (Fig. 45.3). Asymmetry between sides, which was marked initially, became less prominent with disease progression. In contrast, the anterior–posterior gradient of dopamine dysfunction was maintained throughout the course of illness, suggesting that the rate of decline did not differ between subregions (41). Progression has also been evaluated using metabolic networks, which have shown that the spatial topography of the PDRP remains unchanged over time, and the degree of PDRP expression increases linearly with disease progression (28).
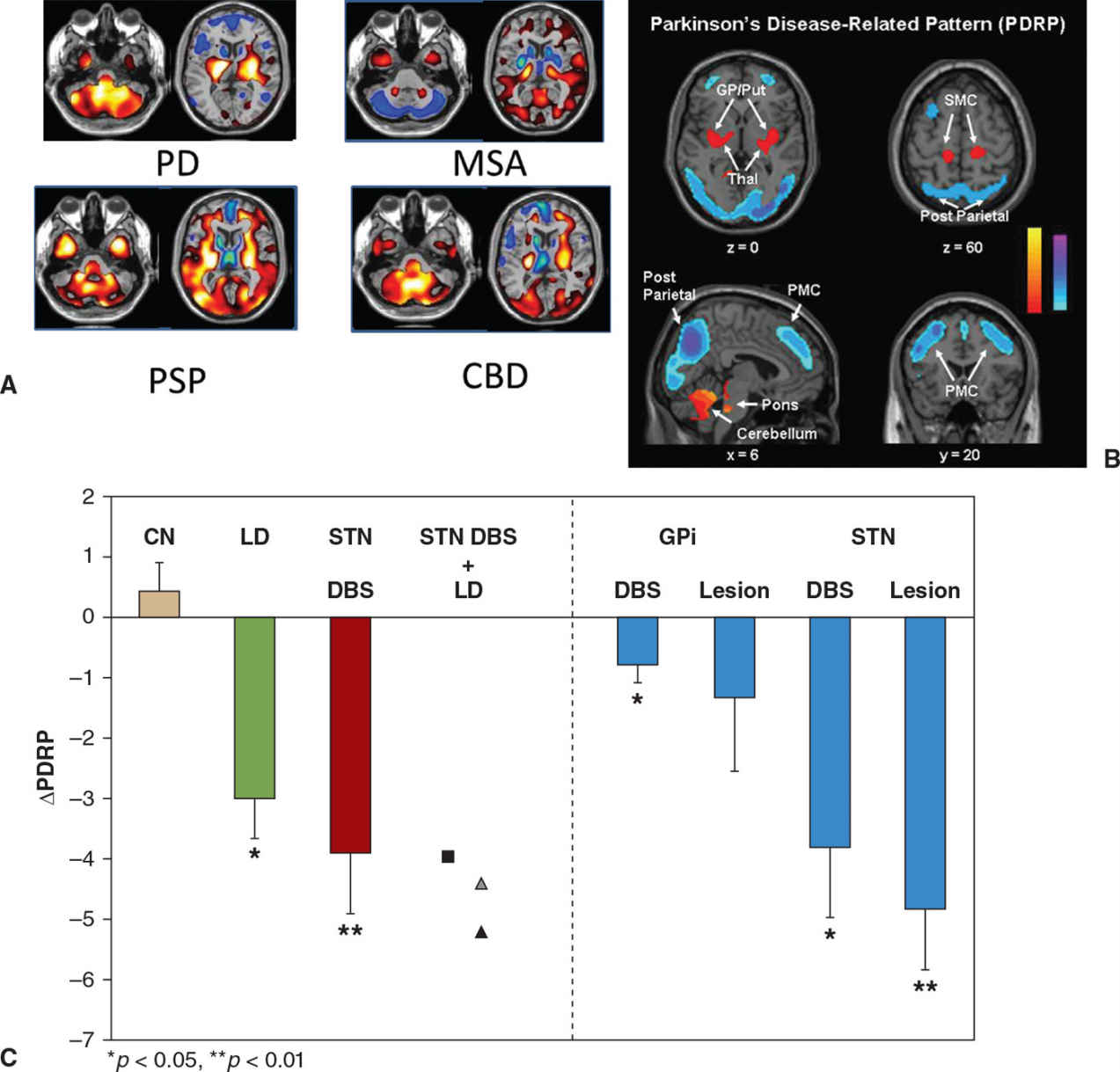
Figure 45.2. (A) Patterns of glucose metabolism differentiate between Parkinson’s disease (PD), multiple system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD). Increased metabolism is shown using ‘hot’ colors and decreased metabolism by ‘winter’ colors. (B) Areas of increased metabolism compared to healthy controls are displayed in yellow or red, while areas of reduced metabolism are displayed in blue. The Parkinson’s disease–related pattern (PDRP) is characterized by increased metabolism in the globus pallidus (GP), putamen, and cerebellum and by reduced metabolism in the posterior parietal and premotor cortex (PMC). (C) The PDRP is suppressed by treatments known to reduce parkinsonian manifestations, including levodopa (LD), subthalamic nucleus deep brain stimulation (STN DBS), and lesions of either the internal globus pallidus (GPi) or STN in contrast to test-retest control subjects (CN). (A from Eckert T, et al. FDG PET in the differential diagnosis of parkinsonian disorders. Neuroimage 2005;26:912–921. B and C from Asanuma K, et al. Network modulation in the treatment of Parkinson’s disease. Brain 2006;129:2667–2678.)
INFLAMMATION
There has been growing interest in the role of inflammation in neurodegenerative disorders such as PD (42). Activated microglia are an important biomarker for neuroinflammation (42). Animal models have demonstrated that microglial activation precedes neurodegeneration (43), and postmortem studies have shown that increased microglial activation is widespread in PD (44). Microglial activation has been studied with the help of 11C-(R)-1-(2-chlorophenyl)-N-methyl-N-(1-methylpropyl)-3-isoquinoline carboxamide (11C-(R)-PK 11195), and other newer agents, which are ligands for the translocator protein (TSPO, previously known as the peripheral benzodiazepine receptor), found on activated microglia. These studies have provided contrasting results. In one study, increased microglial activation was confined to the substantia nigra, and correlated with disease severity and degree of dopaminergic neuronal loss (45). However, in another study, changes were more widespread and did not correlate with either dopamine cell loss or disease severity (46). A third study did not find significant differences between PD and controls (47). Although the newer agents possess a better signal to noise ratio, their affinity to TSPO varies between individuals, based on a polymorphism of the TSPO gene, thereby complicating the interpretation of the imaging findings and limiting the number of subjects in whom such studies can be performed (48).
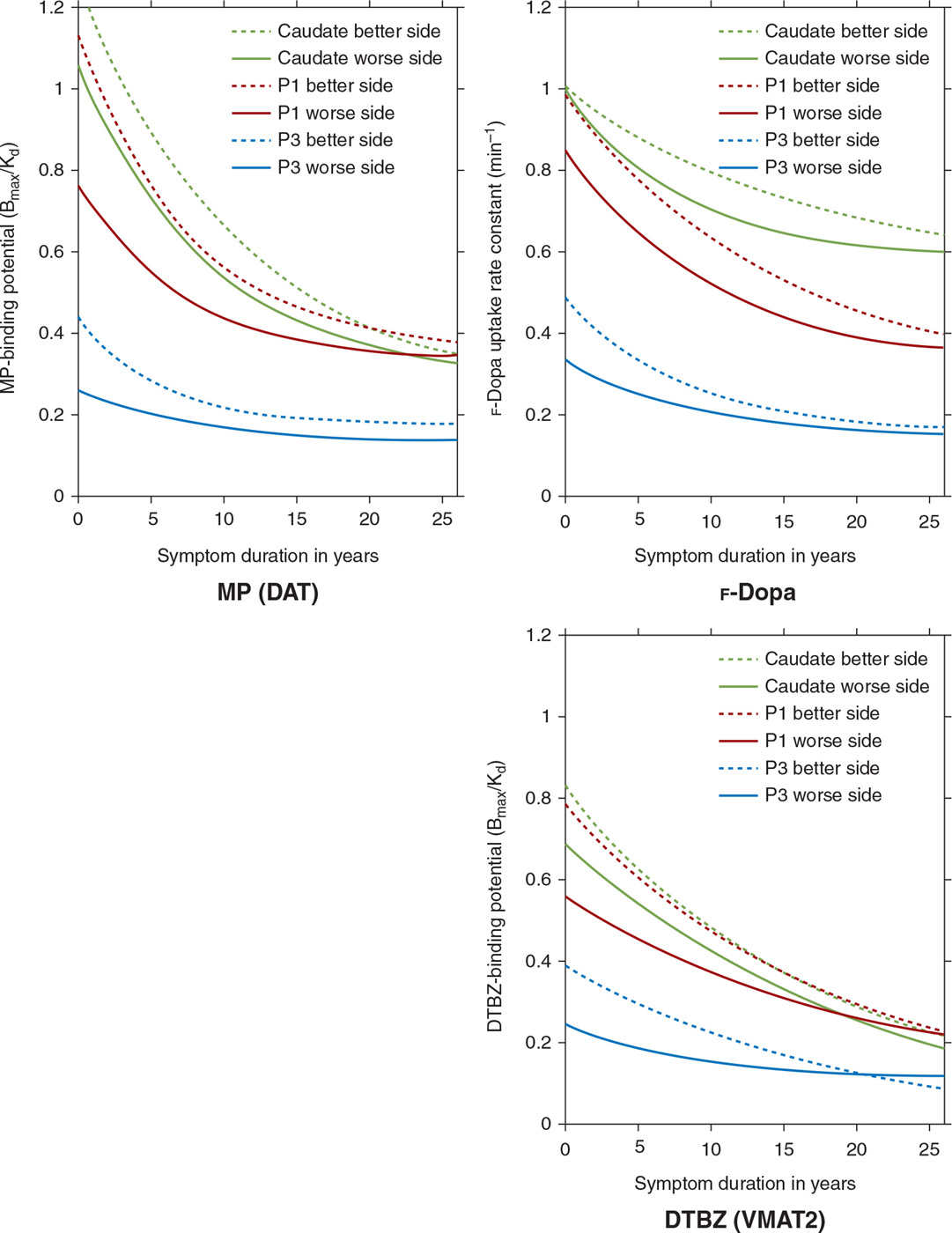
Figure 45.3. Multitracer longitudinal studies of dopaminergic function in a cohort of 78 patients with sporadic PD show an exponential pattern of decline with a rostrocaudal gradient of function in which the caudate remains least affected over the course of the illness, while the posterior putamen (P3) is maximally affected and the anterior putamen (P1) is affected to an intermediate degree. (Data from Nandhagopal R, et al. Longitudinal progression of sporadic Parkinson’s disease: a multitracer positron emission tomography study. Brain 2009;132:2970–2979.)
ABNORMAL PROTEIN DEPOSITION
In PD, α-synuclein is misfolded and aggregates abnormally in cell bodies and neurites, and is abundant in Lewy bodies. The ability to image α-synuclein deposition would be helpful in the diagnosis of PD, and would potentially provide an important tool to study disease progression, as well as response to disease-modifying therapies in the future. Unfortunately, the development of such imaging techniques has been more challenging than in AD, where imaging of amyloid Aβ deposition using Pittsburgh compound (PiB) has opened new avenues of research. The difficulties arise from the low levels of α-synuclein deposition, its intracellular location, and the poor selectivity of agents for α-synuclein versus other misfolded proteins. 11C- and 18F-labeled benzoxazole compounds which bind to α-synuclein have been developed, but they also bind to β-amyloid, and have not been widely used in the study of PD (49,50). 123I-SIL23 has also been shown to bind to α-synuclein fibrils in vivo as well as in vitro using mouse models; however, this ligand also binds to amyloid Aβ and tau, albeit to a lesser degree (51). There is evidence of amyloid Aβ deposition in the cortex of patients with Lewy body disorders such as PD, Parkinson’s disease with dementia (PDD), and dementia with Lewy bodies (DLB). Imaging with PiB demonstrated increased binding in these disorders, especially those with DLB, and to a lesser and more variable extent in PDD. In PD without dementia, increased PiB binding has not been seen (52). Aβ and α-synuclein may have a synergistic effect in the development of cognitive impairment in PD and related disorders (52).
IMAGING THE COMPLICATIONS OF PD
Fluctuations and Dyskinesia
Functional imaging has provided us with insights into the pathophysiology of fluctuations and dyskinesia in PD. Dyskinesia in PD are a complication of therapy, and are generally due to medical treatment with L-dopa, termed “levodopa-induced dyskinesia” (LID), but have also been seen in patients who received striatal fetal mesenchymal stem cell transplants, termed “graft-induced dyskinesia” (GID). Several hypotheses have been considered to explain their occurrence.
Presynaptic dopaminergic system abnormalities are thought to play a role in the development of dyskinesia based on the following evidence. PD patients with motor fluctuations (MFs) were found to have decreased 18F-FDOPA activity compared to those with a stable response to medications (53). Those with dyskinesia were more likely to have reduced DAT function compared to patients without dyskinesia, even after correcting for the degree of dopamine nerve terminal loss (54). This would be in keeping with increased dopamine turnover, whereby dopamine undergoes enzymatic degradation in the synaptic cleft rather than being reutilized following reuptake into the presynaptic terminal (55). This is further supported by studies which estimate synaptic dopamine concentration based on the binding of 11C-raclopride in the striatum, which have shown that in patients with MF, synaptic dopamine levels peak earlier and decrease faster than in patients with a stable response to medications (56). Using similar techniques, it has been shown that patients with peak-dose dyskinesia have increased synaptic dopamine concentrations, which correlate with the severity of dyskinesia (57,58) (Fig. 45.4). Animal studies indicate that repeated pulsatile stimulation of denervated dopamine receptors leads to changes downstream to the receptors. In keeping with the studies in animal models, in vivo studies with PET have failed to demonstrate any consistent relationship between motor complications and dopamine-receptor binding (59).
Recent studies have shown that the serotonergic system may play a role in the development of dyskinesia. Serotonergic neurons have been shown to convert L-dopa into dopamine, and release it in into the synaptic cleft. However, this occurs in a nonphysiologic manner, as these neurons lack mechanisms for autoregulatory feedback. In advanced PD, since most dopaminergic neurons are lost, unregulated dopamine release via relatively intact serotonergic terminals has been postulated to be responsible for dyskinesia. This has been demonstrated in a PET study using 11C-DASB and 11C-raclopride, wherein there was relative preservation of serotonergic terminals, and both increased synaptic concentrations of dopamine following L-dopa, as well as dyskinesia were attenuated by preadministering a 5-HT1A (autoreceptor) agonist (18).
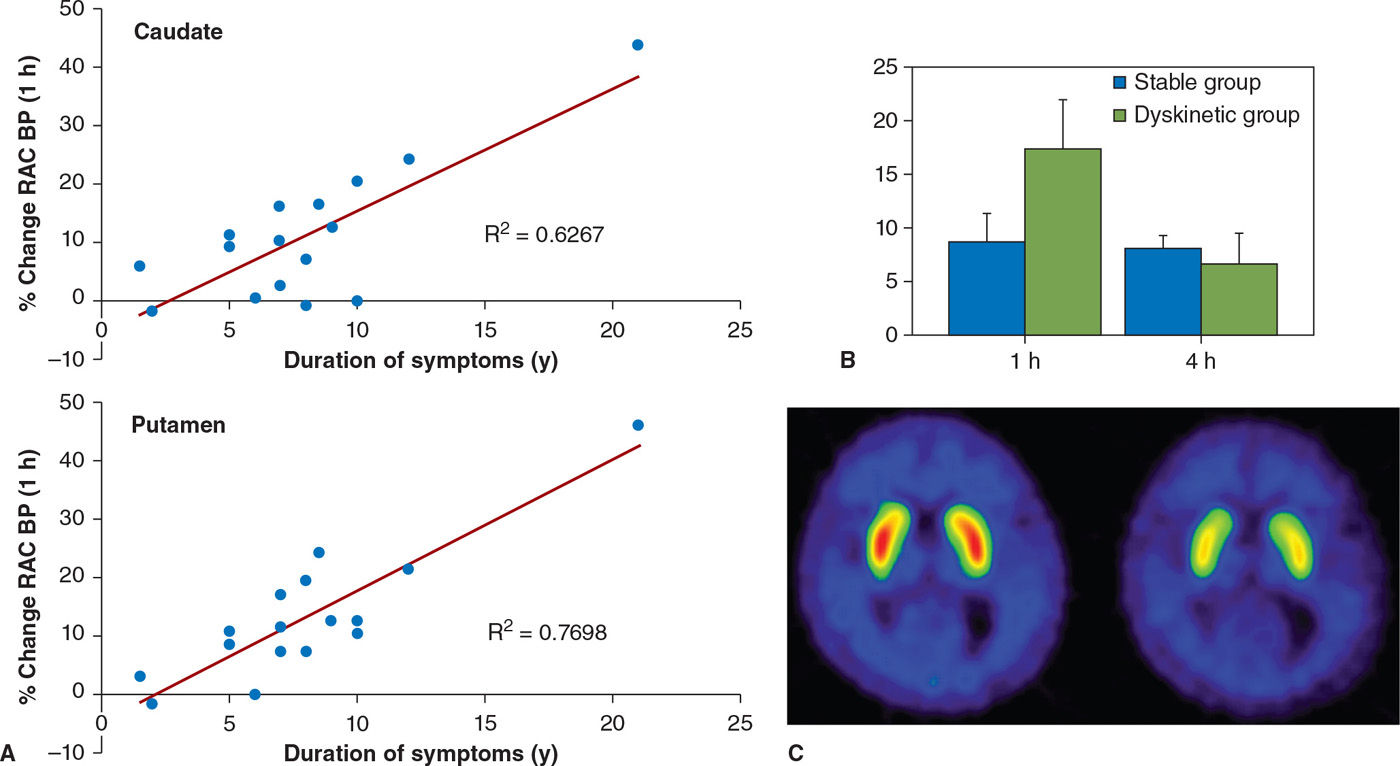
Figure 45.4. (A) The magnitude of dopamine release following oral levodopa (displayed as % change in raclopride binding) increases with disease duration. (B) In patients with levodopa-induced dyskinesias, there is a marked increase in dopamine release 1 hour following levodopa, that is poorly sustained. In patients with a stable response to levodopa, there is a modest release of dopamine following levodopa, which is sustained for at least 4 hours. (C) shows raclopride binding in a patient with dyskinesias at baseline and 1 hour following oral levodopa. The marked reduction in binding following levodopa is indicative of dopamine release. (From de la Fuente-Fernandez R, et al. Levodopa-induced changes in synaptic dopamine levels increase with progression of Parkinson’s disease: implications for dyskinesias. Brain 2004;127:2747–2754.)
PET studies have shown increased glutamatergic activity in the basal ganglia and precentral gyrus in patients with dyskinesia while “on” L-dopa, using 11C-CNS 51619, a marker of activated glutamate receptor ion channels (60). A PET study investigating the role of the opioid system in dyskinesia has found that the binding of 11C-diprenorphine, a nonselective opioid antagonist, is reduced in the striatum and thalamus, and increased in the prefrontal cortex, among patients with LID (25). Adenosine 2A receptors have been shown to be increased in the striatum of PD patients with LID using two different ligands (26,61).
Impulse-Control Disorders
Impulse-control disorders (ICD) are behavioral abnormalities which arise due to dysfunction in the mesolimbic dopaminergic pathways, and in PD, are most commonly seen in patients on dopamine agonists. They include disorders such as pathologic gambling, hypersexuality, compulsive shopping, eating, and internet use, as well as dopamine dysregulation syndrome (DDS, compulsive abuse of dopaminergic medication) and punding behavior (repetitive non–goal-oriented behavior). In a PET imaging study using 11C-raclopride, PD patients with DDS had an exaggerated release of dopamine in the ventral striatum in response to L-dopa, as compared to PD patients without DDS, despite comparable levels of dopamine release in the dorsal striatum (62). Similar findings of increased dopamine release in the ventral striatum were observed while studying PD patients with pathologic gambling while performing a task with monetary reward (63) or during presentation of visual stimuli associated with the ICD (64) (Fig. 45.5). Functional MRI studies have shown impaired activation of the orbitofrontal cortex in the face of negative outcomes in PD patients on dopamine agonists (65), and have also shown increased activation in the ventral striatum, cingulate, and orbitofrontal cortices when PD patients with hypersexuality were exposed to visual sexual cues, especially when “on” medications (66).
Depression and Anxiety
Although depression and anxiety are common nonmotor manifestations of PD, their neurochemical basis is poorly understood. Depression in PD appears to reflect more than the understandable emotional response to living with a progressive neurologic illness, and may be attributed to impaired dopaminergic transmission in mesolimbic pathways, reduced serotonergic or noradrenergic activity, hence these pathways have been investigated in PD. Imaging of the serotonin transporter has shown reduced activity in the striatum and cortex in patients with PD (67). PET studies with 11C-WAY 100635, which binds to the 5-HT1A receptors, have shown reduced activity in the midbrain (68). However, as mentioned earlier, PD patients with depression have increased serotonin transporter activity, compared to PD patients without depression (19) and normal controls (20). 11C-RTI 32 is a radioligand which binds to both the noradrenaline transporter and the DAT; decreased binding has been demonstrated in the noradrenergic LC, thalamus, ventral striatum, amygdala, and cingulate gyrus in PD patients with depression. Anxiety was also shown to correlate inversely with striatal binding of this radiotracer. These data would suggest that depression in PD is linked to noradrenergic dysfunction in the LC, and preferential dopaminergic denervation in the mesostriatal and mesolimbic projections (14).
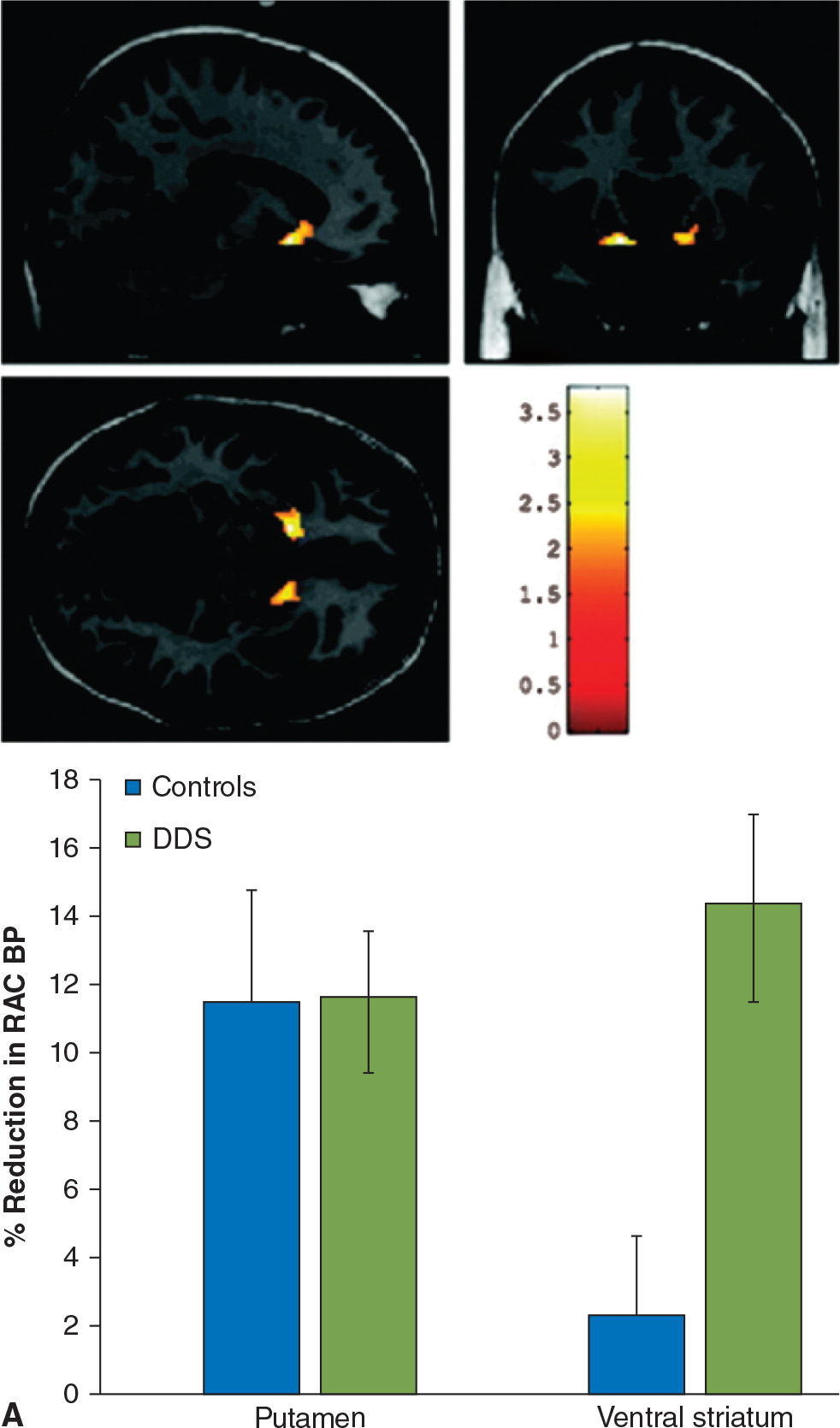
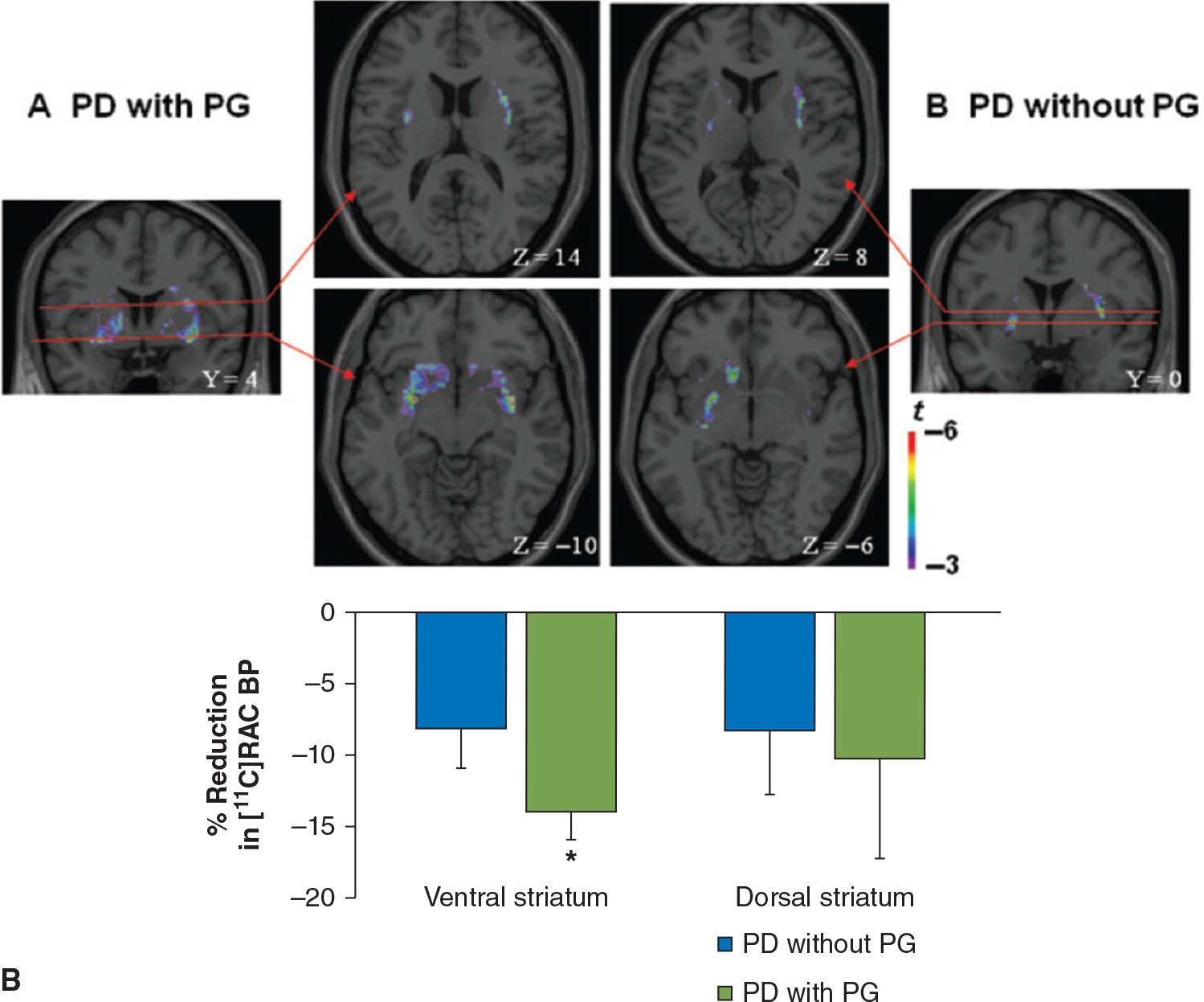
Figure 45.5. (A) In patients with dopamine dysregulation syndrome (DDS), there is a marked increase in dopamine release in the ventral striatum following levodopa administration, not seen in controls (in this case, PD patients without DDS). In the putamen, both groups of patients display a comparable degree of dopamine release following levodopa. (B) Dopamine release is also increased in the ventral striatum of PD patients with pathologic gambling (PG) while they play a card task with monetary reward. (A from Evans AH, et al. Compulsive drug use linked to sensitized ventral striatal dopamine transmission. Annals of Neurology 2006;59:852–858. B from Steeves TD, et al. Increased striatal dopamine release in Parkinsonian patients with pathological gambling: a [11C] raclopride PET study. Brain 2009;132:1376–1385.)
PARKINSON’S DISEASE DEMENTIA AND DEMENTIA WITH LEWY BODIES
Cognitive impairment is common in PD, and is multifactorial in etiology. Dopaminergic deficits in associative striatum and cortical dopaminergic projection regions, cholinergic dysfunction, cortical Lewy body pathology, concomitant Alzheimer’s, and small vessel pathology may all play a role that varies from one individual to another (69). In early PD without dementia, an association between reduced caudate nucleus dopaminergic activity, and cognitive slowing (70), visual memory, immediate and delayed verbal memory, and performance on the Wisconsin card sorting test (a measure of set shifting and cognitive flexibility) has been demonstrated (71,72). In addition, using fMRI, reduced activation of frontostriatal circuits while performing cognitive tasks has been demonstrated in PD, especially in PD patients with mild cognitive impairment (MCI) (73,74). In PDD, imaging with 18F-FDOPA has shown reductions in dopaminergic innervation of the left caudate, bilateral ventral striatum, and anterior cingulate, in comparison to patients without dementia who were matched for age, disease duration, and severity (75). These studies suggest that dopaminergic dysfunction involving the caudate, as well as mesocortical and mesolimbic projections, contributes to cognitive dysfunction in PD and PDD.
In DLB, Lewy body/neurite pathology in the cortex is associated with cognitive dysfunction. However, currently there are no reliable imaging techniques to visualize this type of pathology. In contrast, amyloid Aβ deposition can be imaged, and has been detected commonly in cortical association areas in DLB, less commonly in PDD, but not in PD without cognitive dysfunction (76,77). Although superficially this might suggest that amyloid Aβ plays a significant role in cognitive dysfunction in DLB, it has been shown that the cognitive profile in DLB does not correlate well with the distribution of amyloid Aβ pathology (78). Some authors suggest that amyloid deposition may contribute to dementia, but the severity is determined by the Lewy body pathology. It should be noted that in postmortem tissue from patients with PDD [11C]PiB binds to Aβ, and not to α-synuclein which also has a fibrillar amyloid structure, or neurofibrillary tangles which reflect tau pathology (79). In a more recent study which investigated PD patients with MCI and risk factors for dementia with 11C-PiB, it was found that amyloid deposition was uncommon in PD at risk for dementia; however, neocortical amyloid deposition correlated well with cognitive dysfunction (80).
Cholinergic deficiency has been demonstrated in both PD and PDD using 123I-iodobenzovesamicol SPECT (81), which binds to vesicular acetylcholine transporters, as well as PET using 11C-MP4A and 11C-PMP (82,83), which act as substrates for acetylcholinesterase (Fig. 45.6). In PD, this deficiency is confined to the parietal and occipital regions, whereas it is more widespread in PDD (81) (Fig. 45.6). Moreover, in PDD the global cholinergic deficiency is much greater than in PD (82,83), and correlates with performance on cognitive tests such as the digit span, trail making, and Stroop tests, but not with motor disability or disease duration (83). Muscarinic receptors were studied in PD using 11C-N-methyl-4-piperidyl benzilate (11C-NMPB) PET, which showed increased binding in the frontal cortex; this study also included one patient with PDD, who had the highest levels of binding (84). In contrast, in another study of muscarinic receptors with [123I]iodo-quinuclidinyl-benzilate (123I-QNB) SPECT performed on PDD subjects, levels were found to be low, attributed to cortical Lewy body pathology (85).
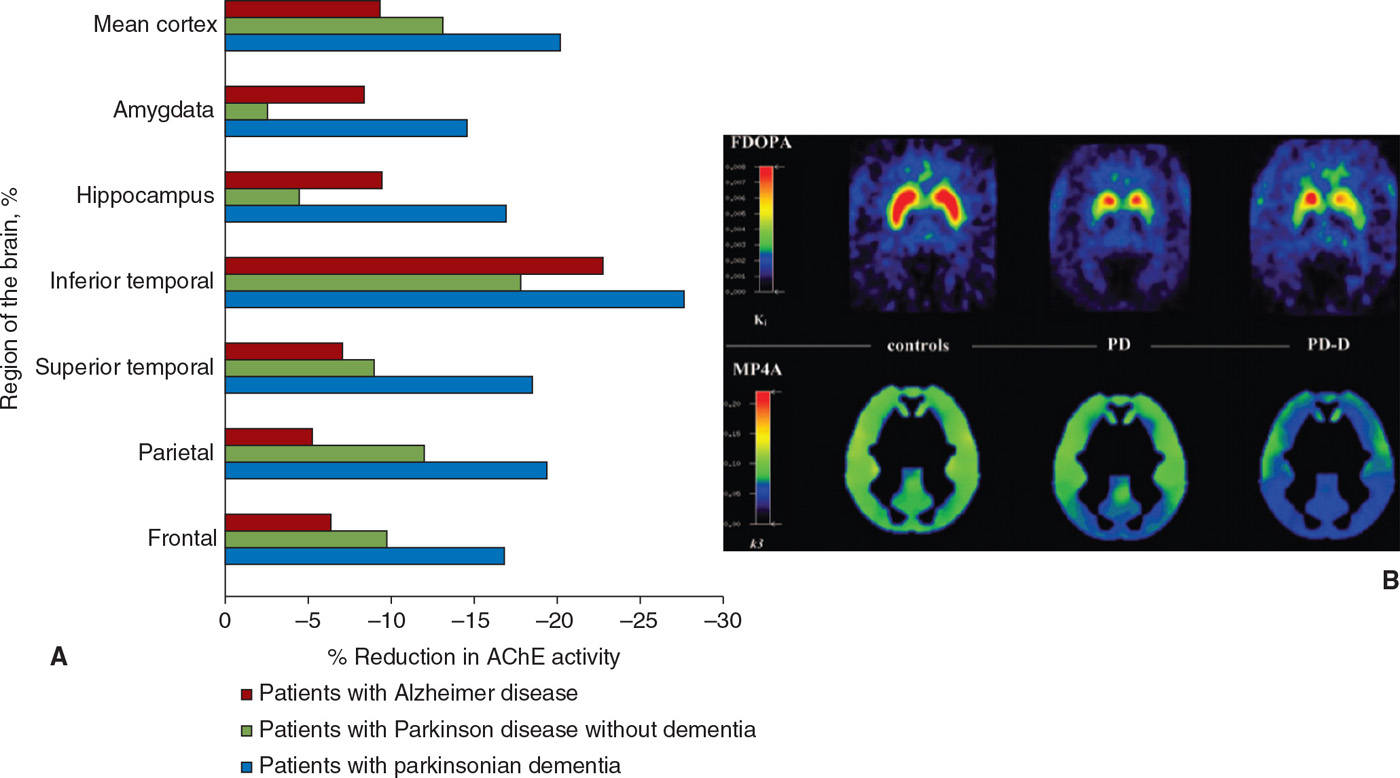
Figure 45.6. (A) Cholinergic innervation, assessed using tracers for cholinesterase activity, is reduced in multiple regions in Alzheimer disease and to an even greater extent in PD with dementia. (B) A greater degree of cholinergic impairment is seen in PD with dementia (PD-D) as compared to PD using MP4A, while the degree of striatal dopaminergic denervation measured using FDOPA is comparable. (A from Bohnen NI, et al. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: an in vivo positron emission tomographic study. Arch Neurol 2003;60:1745–1748. B from Hilker R, et al. Dementia in Parkinson disease: functional imaging of cholinergic and dopaminergic pathways. Neurology 2005;65:1716–1722.)








