Gabapentin
David W. Chadwick
Thomas R. Browne
Introduction
Gabapentin is a novel antiepileptic drug (AED) related in structure to the neurotransmitter γ-aminobutyric acid (GABA) (Fig. 1). It was synthesized as a GABA-mimetic agent that could freely cross the blood–brain barrier (BBB). Despite its structural relationship to GABA and its recognized antiepileptic properties, it is doubtful that therapeutic activity results from any form of direct GABAergic effects.
Mechanism of Action
Gabapentin has been shown to prevent seizures in several animal models (Table 1) and in clinical studies. It has a mechanism of action that appears to be different from other AEDs; this topic has been recently reviewed by Sills.35
In vitro, gabapentin does not interact with neuronal sodium channels or L-type calcium (Ca) channels.39 Gabapentin is inactive at concentrations up to 100 μmol in standardized tests for interaction at GABAA, GABAB, benzodiazepine, glutamate, N-methyl-D-aspartate (NMDA), quisqualate, kainate, glycine, MK-801, and strychnine-insensitive glycine receptors. In addition, many other neurotransmitter receptors have been screened, including A1 and A3 adenosine receptors; α1, α2, and β-adrenergic receptors; D1 and D2 dopamine receptors; histamine1 receptors; S1 and S2 serotonic receptors; M1, M2, and nicotinic acetylcholine receptors; μ-, δ-, κ-, and σ-opiate receptors; leukotriene B4, D4, and thromboxane A2 receptors; phorbol ester dibutyrate receptors; and binding sites on Ca channels labeled by nifedipine and diltiazem and on sodium channels labeled by batrachotoxinin.39 These negative results support the idea of a novel mechanism of antiepileptic action for gabapentin.
Although gabapentin does penetrate the BBB effectively,47 and has antiepileptic properties in vivo,39 it does not interact with GABAA receptors, nor is it converted to GABA or a GABA agonist. Early evidence did not suggest that gabapentin is an inhibitor of GABA uptake nor of degradation by GABA transaminase at relevant concentrations. However, it can increase GABA accumulation in rat brain, by increasing GABA synthesis41 and preventing metabolism.20 In human brain slices16 and in the intact brain, as measured by magnetic resonance (MR) spectroscopy,32 it may indeed have modest effects in increasing brain concentrations. It may also have some effects on the GAT-1 GABA transporter system.51 The degree to which this may contribute to its antiepileptic effects remains uncertain.
It may also possess some baclofen-like effects at the presynaptic GABAB receptor. It can reduce potassium-evoked Ca influx via voltage-gated channels in a pituitary cell line expressing functional subunits of the receptor, an effect reversed by other selective GABAB antagonists.8,30 This may enable gabapentin to selectively reduce excitatory neurotransmitter release with relative sparing on inhibitory neurotransmission. This is despite any evidence that gabapentin can interact with the GABAB receptor.39
Specific binding of gabapentin is highest in the superficial layers of neocortex and dendritic layers of hippocampus, with low levels of binding in the white matter and brainstem.19 This binding site, which doesn’t bind any other conventional AED, has now been identified as the α2δ subunit of the voltage-gated Ca channel.18,49 It appears that two of four isoforms of this subunit are capable of binding gabapentin and its structural analogs.40 There is now a general consensus that this is the site of action responsible for the great majority of gabapentin’s pharmacologic antiseizure activity. It does not appear to act as an inhibitor of any conventional subtype of voltage-gated Ca channel, but rather acts as a selective blocker of channels that contain the α2δ-1 subunit.11
Pharmacologic Fundamentals
Gabapentin is absorbed via the L-amino acid transport system in the proximal small intestine. Absorption is rapid, with maximum plasma concentrations occurring 2 to 3 hours after oral administration.46 Gabapentin appears to move across the gut and into the blood by a saturable transport mechanism competitively inhibited by L-leucine36 (the amino acid transporter known as system L). Experiments with the system L transporter in cultured Chinese hamster ovary tumor cells also support the notion that gabapentin competes with L-leucine, L-valine, and L-phenylalanine and is itself a substrate for transport by system L. This may explain why gabapentin penetrates into the brain: The amino acid structure of gabapentin and the very high hydrophilicity of the compound would otherwise reduce its access across biologic membranes. The time of peak antiepileptic action with gabapentin is delayed about 2 hours after intravenous administration, past the time of peak drug concentration in either the blood plasma or the brain interstitial space.50 Structure activity studies indicate that activity in seizure models correlate with affinity for this transporter system,37,38 although it remains doubtful that the transporter has anything other than an indirect relationship to gabapentin’s ultimate antiseizure activity.
Absorption is dose dependent due to saturation of the L-amino acid transport system. The bioavailability of gabapentin decreases from 0.54 ± 0.11% at a dosing rate of 400 mg three times daily, to 0.35 ± 0.07% at a dosing rate of 1,200 mg three times daily. The bioavailability of gabapentin is not affected by food.
Gabapentin is not bound to plasma proteins to any significant degree. Gabapentin is transported from plasma across the BBB to the brain via the L-amino acid transport system. Gabapentin concentration in a single specimen of human brain was 80% of plasma concentration, confirming animal distribution studies.31
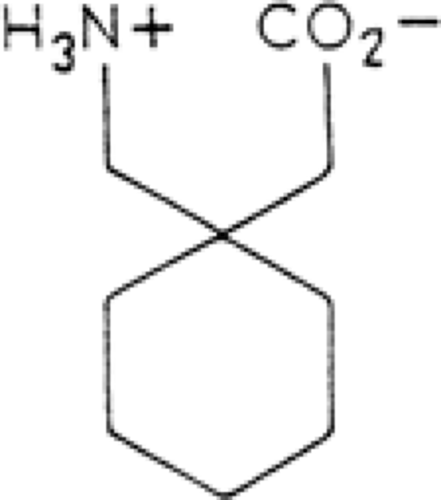 FIGURE 1. Structure of gabapentin. |
Table 1 Activity of gabapentin in animal models of seizures | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Gabapentin is not metabolized.46 It is excreted unchanged in urine, with a renal clearance approximately equal to
total clearance (120–130 mL/min) and an elimination half-life of 5 to 7 hours. The clearance and elimination half-life of gabapentin are not altered by increased dosing rates or by chronic administration.44 The clearance of gabapentin is decreased in the presence of renal disease, requiring a decrease in dosing rate for persons having a creatinine clearance rate of less than 60 mL/min. This mechanism probably is responsible for the observed age-related decrease of gabapentin clearance in later life. Pharmacokinetic studies in children show an increased clearance in children under the age of 5 years.
total clearance (120–130 mL/min) and an elimination half-life of 5 to 7 hours. The clearance and elimination half-life of gabapentin are not altered by increased dosing rates or by chronic administration.44 The clearance of gabapentin is decreased in the presence of renal disease, requiring a decrease in dosing rate for persons having a creatinine clearance rate of less than 60 mL/min. This mechanism probably is responsible for the observed age-related decrease of gabapentin clearance in later life. Pharmacokinetic studies in children show an increased clearance in children under the age of 5 years.
A series of studies have been completed to detect possible interactions between gabapentin and other drugs. These have been conducted either in patients receiving antiepileptic monotherapy or in healthy subjects. An interaction between gabapentin and one of the established AEDs would influence the design and interpretation of phase II and III efficacy studies, and early evaluation of this possibility was important. Interactions in both directions have been sought: that is, an effect of gabapentin on the kinetics of the established drugs and vice versa. In addition, selected potential interactions have been examined with the following compounds:
An aluminum/magnesium hydroxide antacid, because these preparations reduce the absorption of some drugs
Cimetidine, which is a known inhibitor of the oxidative, drug-metabolizing system in the liver
Probenecid, which can inhibit renal tubular secretion of acidic drugs
The components of the combined oral contraceptive pill, the metabolism of which is enhanced by enzyme-inducing AEDs
No detectable interaction occurs between gabapentin and the four AEDs tested (carbamazepine, phenytoin, phenobarbital, valproate).33 This would be expected of a drug such as gabapentin, which is renally eliminated and does not induce hepatic microsomal enzymes.
The aluminum/magnesium antacid Maalox TC (Rhone-Poulenc Rorer) caused a slight reduction in the absorption of gabapentin, but no more than 20%, which is probably not of clinical significance.13 Cimetidine decreased glomerular filtration and caused a 12% decrease in the renal clearance of gabapentin; but again this is unlikely to be of clinical importance. Probenecid, however, did not affect renal clearance, indicating that gabapentin does not undergo renal tubular secretion by the pathway blocked by probenecid. Finally, no effect of gabapentin was seen on the kinetics of single doses of norethynodrel and ethinylestradiol, which is predictive of a lack of interference with oral contraception.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


