Fig. 1
rAAV2/5 efficiently transduces neurons and produces equal protein expression in the young and aged rat hippocampus (HP). (a–d) Young adult (a and c, 6 months, n = 3) and old aged (b and d, 20 months, n = 3) Fischer 344 rats were injected with 2 μl (titer 1 × 1013 vg/ml, 0.3 μl/min infusion rate) rAAV-GFP (a and b) or rAAV-human tau (c and d) into the dorsal HP (−3.8 mm AP and +2 mm ML from bregma, and −2.6 mm DV from the dura) using pulled glass syringe tips attached to a Hamilton gastight syringe (#7653-01). In all conditions, rAAVs produced widespread transduction of hippocampal neurons. Endogenous GFP signal was used for imaging GFP injected animals, while Tau12 antibody (1:40,000; [144]), a human tau-specific monoclonal antibody, was used to label human tau in rAAV-tau injected animals. Our group has observed efficient transduction of all neuron types in the HP with rAAV2/5 (e.g., CA pyramidal neurons and dentate granule cells). Scale bar = 400 μm. (e) Lysates of the dorsal HP were run on a western blot to quantify the amount of rAAV-derived proteins (e.g., GFP and human tau, n = 3/group). Blots were probed with GFP antibody (Abcam, ab290, 1:3,000), Tau12 (1:200,000), and two loading controls βIII-tubulin antibody (Tuj1, 1:10,000; [145]) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, Cell Signaling, 5174, 1:2,000). The signal intensity for each band was quantified using the Licor Image Studio software and signal intensities for GFP or tau are expressed as a ratio to tubulin signal intensities (similar results were obtained when normalized to GAPDH signal). No aging-related differences were observed with either GFP levels (unpaired t-test, p = 0.93) or human tau levels (unpaired t-test, p = 0.46). Immunofluorescence and Licor immunoblotting were performed similar to previously published methods [146]
In addition to great temporal control, viral vectors provide control over the spatial expression of the transduced genes. AD and other tauopathies are characterized by the degeneration and pathological accumulation of proteins in specific brain regions. For example, the EC and HP are primary affected areas in AD, while other tauopathies involve degeneration in the frontal and temporal cortices, as well as the basal ganglia, brainstem and cerebellum. Viral vector s allow researchers to stereotaxically inject viruses in relatively discrete brain regions of interest. For example, direct injection of rAAVs into the rat HP or EC results in efficient transduction of neurons (Fig. 2). Furthermore, unilateral injections allow the contralateral half of the brain to serve as a control within the same animal, but the contralateral projections of a specific region must be considered. Another level of specificity can be obtained by using cell-specific promoter systems [86, 87], which can also be used in transgenic models [78, 88]. Finally, the serotypes of AAVs exhibit significant differences in cell tropism allowing for increased specificity of transducing different cell types (e.g., neurons, glia, or both) and specific brain regions [89, 90].
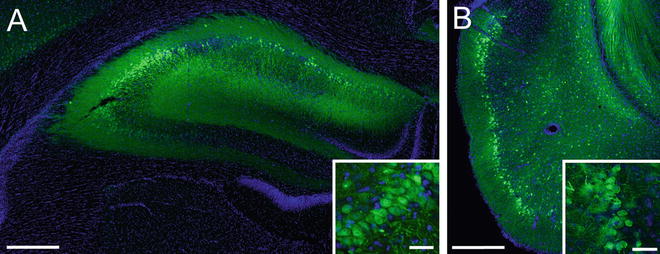
Fig. 2
rAAV2/5 effectively transduces neurons in the hippocampus (HP) and entorhinal cortex (EC) of Fischer 344 rats. (a) A representative image of the dorsal HP of a young adult (6 months) rat transduced with rAAV-GFP (as in Fig. 1, scale bar = 400 μm). The inset illustrates GFP expressing CA3 pyramidal neurons in the HP (scale bar = 50 μm). (b) A representative image of the EC of a young (3 months) rat transduced with rAAV-GFP (titer = 1.9 × 1013, coordinates: −5.8 mm AP, +3.3 mm ML from bregma, −8.8 mm DV from dura, at 17° angle; scale bar = 400 μm). Note that layer II/III EC neurons are well transduced (inset, scale bar = 50 μm); these neurons are particularly susceptible in AD. Endogenous GFP fluorescence was used for imaging transduced cells
Additional advantages of viral vector systems are the relative ease and short timeframe (i.e., a few months) of generating new viral vectors as well as the low cost of production. Typically, the process involves simply cloning the gene of interest into the appropriate rAAV expression plasmid , and then, in the case of many investigators, sending the expression plasmids to a reputable gene therapy core facility for production. Other investigators choose to generate their own viral vectors ([91] and Chapters 7–9). Thus, viral vector models can be adjusted relatively quickly to test new variants of a particular protein or to examine interactions between proteins. Alternately, viruses can introduce RNA -based inhibitors of gene expression (e.g., shRNA or microRNAs) in a time- and region-specific manner for a relatively simple and selective approach to genetic knockdown [92]. This level of versatility lends itself well to creating numerous novel model systems and easily manipulating the genes/proteins of interest, which is difficult to achieve with other approaches.
5 Potential Caveats with Viral Vector Systems
Every model system has some limitations that are important to acknowledge. rAAVs efficiently transduce cells in the brain , but the level of overexpression can be significant. However, two approaches can help minimize this effect. First, rAAVs can be titered to effectively reduce the copy number per cell, thereby reducing the amount of expression. For example, injection of 2 μl of rAAV-GFP or rAAV-human wild-type tau at a titer of 1 × 1013, 1 × 1012 or 1 × 1011 viral genomes/ml into the dorsal HP of rats (as above) results in a dose-response of expression levels. The regional HP signal for GFP or human tau levels (using Licor Odyssey densitometry analysis) decreased linearly (GFP − r 2 = 0.92, p < 0.001; human tau − r 2 = 0.93 p < 0.001; Fig. 3a, b). Analysis of tau fluorescence levels in individual cells found that the average level of tau expression was approximately ninefold, fourfold or twofold over endogenous tau levels at titers of 1 × 1013, 1 × 1012 or 1 × 1011, respectively (Fig. 3c). Second, regulatable promoter systems, such as tetracycline induction systems, provide an effective level of control over transgene expression [85] but leakiness is always a concern with these approaches (as with regulatable transgenic animals). Nonetheless, it is imperative that investigators assess the level of overexpression when using any model system.
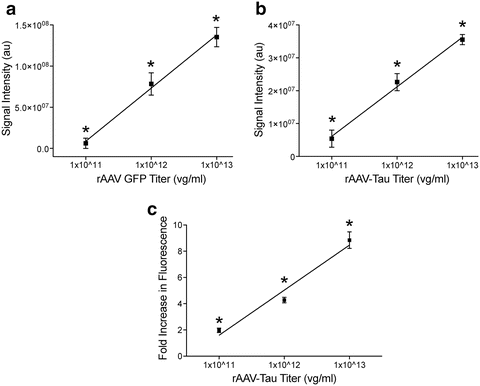
Fig. 3
rAAV2/5 titer-dependent changes in levels of transgene expression and fold-increase in expression are linear in the rat hippocampus (HP). (a) Young Fischer 344 rats (3 months, n = 3/group) were injected in the dorsal HP with rAAV-GFP at 1 × 1011, 1 × 1012, or 1 × 1013 vg/ml (as described in Fig. 1). After 1 month of expression, the tissue was processed for GFP immunofluorescence using GFP antibody (1:28,000) and Licor goat anti-rabbit IRDye 680 (1:500). The fluorescence signal intensity was quantified using Licor Odyssey densitometry analysis in the dorsal hippocampus. Increasing the titer produced a linear increase (r 2 = 0.92, p < 0.001) in regional GFP signal intensity. Each increase in titer produces a significant increase in signal (one-way ANOVA, *p < 0.05 vs. other groups). (b) Using the same parameters, animals (n = 3/group) were injected with rAAV-human tau at the three titer doses. Again, a linear increase (r 2 = 0.93, p < 0.001) in regional human tau (Tau12; 1:40,000, Licor goat anti-mouse 680) signal intensity was observed with increasing titers (using Licor densitometry). Each increase in titer produces a significant increase in signal (one-way ANOVA, *p < 0.05 vs. other groups). (c) Sections from rAAV-human tau animals were stained with dual immunofluorescence for Tau5 (a mouse monoclonal pan-tau antibody that labels human and rodent tau equally, 1:50,000; [147]) and a rabbit anti-flag tag antibody (Sigma, F7425, 1:2,000) to obtain estimations of the level of tau overexpression on an individual cell basis. The human tau proteins are flag tagged, which is a small epitope tag (DYKDDDDK), to facilitate distinction of the human tau from endogenous rat tau. Transduced cells were identified with the flag tag staining and levels of Tau5 fluorescence were measured in the cytoplasm of individual neurons. Normal physiological levels of endogenous rat tau were measured in non-transduced cells (i.e., flag tag negative) in the contralateral hemisphere. The data are presented as the fold-increase in fluorescence intensity (i.e., tau protein levels) from non-transduced cells. As with the regional analyses, a linear increase in fluorescence was observed with increasing titers (r 2 = 0.93, p < 0.001). The range of the fold-increase in expression Fig. 3 (continued) within individual cells was 1–3 for 1 × 1011, 2–14 for 1 × 1012, and 2–17 for 1 × 1013. Each increase in titer produces a significant increase in signal (one-way ANOVA, *p < 0.05 vs. other groups). Of note, this method can only approximate relative expression levels and does not allow precise quantitative measures of protein levels in each cell. Also, methods that do not allow single-cell analysis (e.g., western blotting or ELISAs) are inappropriate for estimating overexpression levels as the samples contain significant quantities of non-transduced cells/tissue. Finally, the reduction in viral titer reduces expression levels, but also decreases the amount of transduced neurons. Thus, one must balance the level of overexpression with transducing an adequate number of cells
Another potential caveat of rAAVs is the lack of absolute specificity in cell-type transduction. Preferential tropism of certain cell types does occur with different rAAV serotypes, but some tropism for other cell types exists as well. For example, rAAV 2/5 is typically described as a neuron-specific serotype , however, a low level of glial transduction is often observed. One mechanism to overcome this is to utilize cell-type specific promoters. Viruses that utilize neuron-specific (e.g., synapsin I) or glial-specific (e.g., glial fibrillary acidic protein) promoters can help address this caveat [87, 93]. Moreover, cell phenotype-specific markers (e.g., tyrosine hydroxylase promoter for monoaminergic neurons) can increase specificity to neuronal subpopulations. As the use of rAAVs becomes more common in the AD and tauopathy field, it will be important to pursue these more refined approaches.
Finally, delivery of rAAV into the brain requires surgical interventions, which lead to disruption of the blood–brain barrier and the brain tissues. To reduce disruption, it is advisable to perform the stereotaxic surgery with the aid of a surgical scope and pulled glass syringe tips. Often, this allows minimal disruption of the dura and in some cases the only disruption is the very fine point of the inserting syringe tip. Details on the surgical procedures used for delivering rAAVs intracerebrally can be found in Chapter 14. Additionally, recent evidence suggests that rAAV 2/9 can effectively enter the brain after a peripheral injection, thereby, circumventing the need for surgery [94].
6 AD and Other Tauopathy Models Using rAAV Tau
The initial rAAV tauopathy models used rAAV-2/2 to express full-length human tau with the P301L mutation in the brains of mice and rats [43, 95]. rAAV-P301L tau was injected into the basal forebrain of adult rats leading to detection of increased levels of the tau protein in the brain for at least 8 months after injection. Within 3–4 weeks hyperphosphorylated tau was found and aggregates resembling NFTs were present although in relatively low numbers. These studies provided the first proof-of-principle that injection of rAAV-P301L tau could result in persistent expression of tau protein and tau aggregation in rodents.
Injection of rAAV-wild-type or triple-mutant APP led to some Aβ plaques but no overt neurodegeneration. In contrast, rAAV-wild-type tau or P301L tau caused significant neurodegeneration of HP pyramidal neurons, but tangles were not present [96]. The tau-induced neurodegeneration was dependent on the microtubule-binding domains, as a truncated version of the protein did not induce toxicity. Interestingly, P301L tau apparently induced cell cycle reentry as indicated by an increase in the presence of cell cycle markers (e.g., cyclins B1 and D2, proliferating cell nuclear antigen, phosphorylated retinoblastoma protein). However, the neurodegenerative effects of injecting rAAV-wild-type tau into the dorsal HP were not replicated in another study using rats [97]. Differences in species, age at rAAV delivery, duration of expression, viral titer and/or neuroinflammatory response may help to explain the discordant findings between these studies.
In a recent experiment, our group used rAAV2/5 to express GFP (used as a control rAAV) or wild-type human tau (longest 4R isoform of 441 amino acids) in the dorsal HP of old Fischer 344 rats (20 months) for 1 month (Fig. 4). Both constructs were well expressed by HP neurons (CA1 pyramidal neurons depicted). Notably, there were clear signs of axonal degeneration, such as fragmentation of tau expressing HP axons in the fimbria, while GFP expressing axons appeared normal. Moreover, the pattern of HP staining with SMI312 antibody, an axon-specific neurofilament antibody cocktail, showed an apparent reduction in axons and the remaining axons appeared dystrophic and fragmented. These data support the usefulness of using rAAV approaches to model early pathological changes such as axonal degeneration and highlight the ability to use aged rats in such studies.
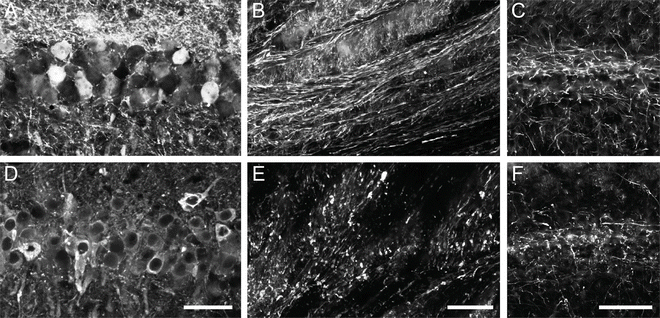
Fig. 4
Expression of human tau using rAAV2/5 causes axonal degeneration in the aged rat hippocampus (HP). Aged Fischer 344 rats (20 months, n = 3/group) were injected in the dorsal HP (as described in Fig. 1) with either rAAV-GFP (a–c) or rAAV-human tau (d–f). (a) and (d) The CA1 pyramidal neurons were well transduced in both cases (a—GFP fluorescence and d—Tau12 immunofluorescence). (b) and (e) Examination of the HP-derived axons in the fimbria revealed that GFP+ axons appeared normal and continuous (b), while human tau + axons showed clear signs of degeneration (e.g., fragmentation and dystrophy). (c) and (f) SMI312, an axon-specific neurofilament monoclonal antibody cocktail (Covance, SMI-312R, 1:10,000) revealed an apparent reduction and fragmentation of axons in the HP (CA1 region depicted) of aged animals expressing human tau (f) when compared to those expressing GFP (e). Scale bars in d, e, and f = 50 μm. Of note, overt neuron loss in the HP was not assessed in this study
The perforant pathway projects from the EC to the HP and is associated with memory formation [98]. It is also the location of some of the earliest tau pathology in AD and among the regions that undergo significant neurodegeneration [99]. rAAV vectors provide an opportunity to model the early stages of AD and the degeneration associated with the perforant pathway [100]. Expression of rAAV2/9-P301L under the control of a synapsin I promoter produced localized tau expression in the lateral EC and perforant pathway. Multiple forms of phosphorylated and aggregated tau were detected in this region by antibodies and silver staining prior to loss of perforant synapses and neuronal death, seemingly triggered through caspase-mediated apoptosis. HP tau expression was initially present only in the perforant pathway projections but was later observed in the dentate granule neurons as well as in target neurons in CA3. This progressive expression was not replicated in rAAV-eGFP controls and may support trans-synaptic spread of tau [100]. We had similar success in efficiently transducing EC neurons in young (3–4 months) Fischer 344 rats using rAAV-2/5. In this experiment, rAAV-wild-type human tau and a phosphomimetic form of AT8 tau (i.e., pseudophosphorylations at S199, S202, and T205, rAAV-psAT8) was injected into the EC of young animals. AT8 tau is a prominent disease-related phosphoepitope of tau and recombinant psAT8 tau significantly impaired anterograde axonal transport as a monomer in the squid axoplasm, an effect not seen with wild-type tau monomers in this assay [19]. Notably, expression of GFP or wild-type tau for 1 month did not appear to alter perforant pathway axons in young animals (Fig. 5a), but AT8 tau expression induced early signs of axonal degeneration (e.g., spheroids and dystrophic axons) in perforant pathway axons (Fig. 5b). One notable caveat of directly injecting the rodent EC is that the syringe tip must pass through the HP (at an angle). Thus, this approach is not well-suited for studying cell-to-cell transfer or the spread of tau because of the likelihood for injected materials to flow back along the needle tract. Nonetheless, the EC can be targeted and overexpression of disease-related forms of tau (i.e., psAT8) appear to induce axonopathy in young animals. Further investigations that study the long-term and aging-related effects of rAAV tau expression in the EC should be pursued.
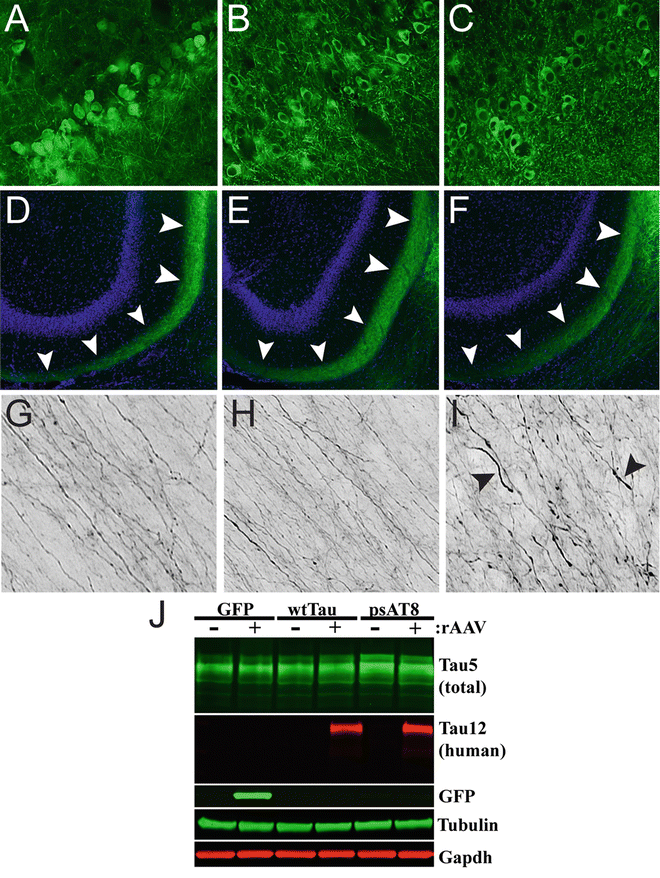
Fig. 5
Expression of a disease-related phosphomimetic form of human tau in the entorhinal cortex (EC) of young rats causes axonal abnormalities in the perforant pathway. Young Fischer 344 rats were injected with rAAV-GFP (a, d, g), rAAV-human tau (b, e, h), or rAAV human tau pseudophosphorylated at the AT8 site (psAT8—S199, S202 and T205) into the EC (c, f, i; as described in Fig. 2; titer = 1.9 × 1013 for all). (a–f) After 1 month, pyramidal neurons of the EC (a–c) were transduced and proteins were present in the outer molecular Fig. 5 (continued) layer of the dentate gyrus (arrowheads in d–f), which is a terminal field of the axonal projections from the EC. (g–i) Notably, axons within the perforant pathway appeared normal in GFP and human tau expressing animals, but in psAT8 tau expressing animals axons appeared dystrophic (arrowheads; i.e., swollen and containing spheroids). Endogenous GFP fluorescence (a, d, g) or Tau12 immunofluorescence (b, c, e, f, h, i) were used to identify rAAV-GFP or rAAV-human tau and rAAV-psAT8, respectively. (j) Western blot was used to confirm the expression of rAAV-GFP, rAAV-human tau and rAAV-AT8 tau in the EC (+ samples are the injected side, − samples are the contralateral HP). Immunoblotting antibodies were used as described in Fig. 1, and Tau5 was used at 1:100,000. Overt neurodegeneration was not assessed in this study
7 Tau in the Substantia Nigra
The presence of tau pathology and neuronal loss is observed in the SN in many tauopathies, including PSP, CBD, FTDP-17, as well as AD (around Braak Stage VI) and Parkinson’s disease [101–104]. rAAV2/2-mediated expression of wild-type or P301L tau in the SN of rat caused early axonal degeneration that was followed by neuronal loss [105]. Accordingly, the animals displayed motor deficits and a directional bias in amphetamine-induced rotational behavior. These behavioral changes occurred in the absence of large numbers of NFT-like inclusions, indicating that tangle-like inclusions may not be required for neuronal dysfunction. In another study, a number of different rAAV serotypes (i.e., rAAV2/2, rAAV2/5, rAAV2/8, rAAV2/9, and rAAV2/10) were tested with P301L tau in the SN [106, 107]. rAAV2/8 P301L induced high tau expression and a loss of approximately 75 % of TH-positive neurons in the SN, while rAAV2/2 and rAAV2/5 induced losses of less than 30 % [106]. rAAV2/9 and rAAV2/10 induced high tau expression and led to the loss of around 90 % of dopaminergic neurons [107]. Interestingly, GFP expression through rAAV2/8 transduction was toxic in the SN at high doses but not in the HP or when expressed through rAAV2/9 [106, 107]. These studies highlight the importance of choosing appropriate control proteins and potential issues with nonspecific toxicity due to dosing effects. Given the losses of 75–90 % of dopaminergic neurons in the SN, this may be of use as a model for some tauopathies that display that particular phenotype, such as PSP, CBD, or FTDP-17 [37, 108, 109].
A study found rAAV2/9 wild-type tau or GFP (at titers of either 2 × 109 or 9 × 109) produced similar levels of expression in young and aged rats in the SN [83, 84]. rAAV-tau expression in the SN caused prolonged microglial activation, neuronal loss and amphetamine-induced rotational behavior in the young and old animals. Interestingly, this effect was enhanced in older rats at lower titers suggesting aged animals are more susceptible to tau toxicity. In fact, expression of wild-type tau with rAAV2/9 induced an upregulation of pro-inflammatory markers and reduced tyrosine hydroxylase mRNA due to loss of dopamine neurons [110], suggesting neuroinflammation might play an important role in tau toxicity in the SN.
8 Mixed Models Using rAAV Tau and Transgenic Mice
In addition to developing novel models, viral transduction can help to improve on existing transgenic models by expressing proteins that introduce or enhance pathology. For example, the PS/APP double transgenic mouse develops amyloid pathology, but only low levels of tau pathology [63]. However, injection of rAAV-P301L tau into the HP of these mice produced moderate tau pathology including early stage tau aggregates and tau-positive neurites surrounding amyloid deposits [95]. Similarly, injection of rAAV2/2-P301L tau into the HP of the triple-mutant APP mouse line, mThy1-hAPP751, resulted in increased phosphorylated tau and tau fibrillar pathology compared to wild-type mice injected with rAAV-P301L tau [58, 111]. Moreover, rAAV-P301L tau induced neuronal loss in the transgenic mice that was not seen in either the wild-type mice or transgenic mice injected with rAAV-GFP, an effect that was attenuated through treatment with a kinase inhibitor [111]. These studies highlight the benefits of combining transgenic mice with rAAV injections to study disease mechanisms and potential therapeutics.
In another series of studies, the TASTPM transgenic mouse line (an APP/PS1 mouse) [112, 113] was injected in the EC with rAAV6 containing wild-type tau, P301S mutant tau, or a modified, enhanced aggregating version of tau named 3PO [114, 115]. The pro-aggregation forms of tau, P301S and 3PO tau, enhanced toxicity around the injection site as well as in CA1 HP neurons and exacerbated microglial activation when compared to wild-type tau. Importantly, the expression of rAAV P301S or 3PO tau resulted in impaired spatial memory. This confirmed previous results demonstrating that transduction of mutant tau into the EC impaired spatial memory of mice [116].
Combining different transgenic lines with rAAV approaches can add novel capability to studying specific processes related to a disease. For example, yellow fluorescent protein transgenic mice were injected with rAAV-P301L tau into the HP, providing a model for tracking axonal degeneration and neuronal loss [117]. In the same study, rAAV-P301L was injected into the HP of transgenic mice harboring the CX3C chemokine receptor 1 gene disrupted with EGFP (produces mice with GFP+ microglia). This study confirmed early degeneration of neuronal processes (i.e., axons and dendrites) prior to overt neuronal degeneration and activation of microglia was detected after the induction of tau expression. These results supported a toxic role for tau prior to the formation of NFT-like aggregates as well as a neurotoxic effect related to microglia.
9 rAAV-Mediated Expression of Other AD- and Dementia-Related Proteins
A number of groups have used rAAVs to model other pathological aspects of AD. For example, rAAVs were used to overexpress Aβ peptides in rodents [118]. Expression of rAAV2/1-BRI-Aβ42, a fusion protein that allows controlled expression and secretion of Aβ peptides [119, 120], in the HP induced the formation of plaques but this was not seen with rAAV2/1-BRI-Aβ40 expression. Another amyloid-based rAAV study generated GFP-tagged versions of Aβ40 and Aβ42, wild-type C100 (a C-terminal fragment of APP containing the Aβ peptides) and a V717F mutant version of C100 and expressed them in the HP and cerebellum of mice using rAAV2/2 [121]. The Aβ42 and V717F C100 led to greater induction of microgliosis and disruption of the blood–brain barrier compared to the Aβ40 forms but did not induce plaque formation, neurodegeneration or astrocyte activation. The use of rAAV technology to express various forms of Aβ and its precursor proteins will continue to be an important method of studying their role in disease.
Similarly, rAAVs were used to induce expression or alteration of other proteins thought to play a role in the mechanisms of AD. Injection of rAAV1-I2NTF and rAAV1-I2CTF, active cleavage products of an inhibitor of protein phosphatase-2A (PP2A), led to phosphorylation and aggregation of tau and Aβ, synaptic defects, and impaired memory in rats [122]. This is an intriguing demonstration of induction of some of the pathological hallmarks of AD without overexpressing mutant forms of tau or APP. Another novel method used rAAV1/2 to express 5-Lipoxygenase (5-LO) in the brains of transgenic APP (Tg2576) or 3×Tg-AD mice resulting in impaired performance in memory tasks and exacerbated Aβ and tau pathology [123, 124]. 5-LO in an enzyme that oxidizes lipids, such as arachidonic acid, releasing several active compounds into the cell and displays increased levels in the HP of AD patients [125].
Some FTDs are characterized by pathological aggregation of TDP-43, a nucleic acid-binding protein, primarily in the frontotemporal cortex and HP (reviewed in Ref. [126]). Overexpression of rAAV2/9-TDP-43 in the SN of rats caused amphetamine-induced asymmetrical rotational behavior, loss of TH-positive neurons, and the development of cytoplasmic TDP-43 aggregates [127]. Similarly, rAAV2/9-TDP-43 expression in the rat HP caused neuronal loss and some memory impairments [97]. Another study took advantage of the flexible nature of rAAVs to address differential toxicities and pathologies due to expression of mutant TDP-43 in neurons and glia of mice [128]. The synapsin 1 and GFAP promoters limited expression of the M337V TDP-43 to neurons and astrocytes, respectively. Neuronal expression resulted in greater TDP-43 aggregation, neuronal loss, and more severe motor defects in the mice. Astrocytic expression also induced neuronal death but only in older animals and without motor defects. These studies demonstrate that rAAV models are a versatile approach to studying the role of pathological proteins as well as the molecular mechanisms of disease, and may help make important distinctions between different cell-specific pathologies.
10 Lentiviral Models
Other models of AD and related neurodegenerative disorders utilize lentiviral vectors, a retrovirus , to express the desired protein as an alternative to rAAV. These differ from rAAVs because they insert themselves into the host genome and have a capacity for larger genetic material. Some of the AD and tauopathy models developed using lentiviral systems are discussed briefly below.
A number of studies used lentiviruses to express wild-type or mutant forms of tau protein in various brain regions of rodents. Lentivirus -mediated expression of P301S tau in the HP of wild-type or transgenic APP23 mice (harboring 2 Swedish APP mutations) was used to examine interactions between tau and Aβ in adult mice [129]. Based on immunostaining, the APP23 transgenic mice displayed increased tau phosphorylation and aggregation compared to the wild-type mice also injected with lenti-P301S. Another group found that P301L tau expression caused an increase in tau phosphorylation, tau aggregation, and activated microglia when compared to wild-type tau expression in the rat motor cortex rats [130]. Lentivirus expression of wild-type or P301L tau in the rat HP produced a progressive appearance of dystrophic and fragmented neurites prior to the formation of NFT-like inclusions that developed over 8 months [131]. Lentiviral expression of wild-type tau, P301L, and Aβ42 led to microglial activation as well as induction of autophagy as fractalkine signaling was increased [132]. This indicates a possible link between the phosphorylated tau, microglia signaling, and induction of autophagy that could be related to apoptotic death of neurons. Finally, lentiviral Aβ42 led to intracellular Aβ accumulation followed by tau hyperphosphorylation that was ameliorated with clearance of the peptide [133].
Lentiviruses were used to express TDP-43, Aβ42, or both together in the motor cortex of rats [134]. Lenti-TDP-43 induced caspase activity, neuroinflammation, and altered the normal processing of APP, which led to increased Aβ42 production, while lenti-Aβ42 induced some caspase-3 activity and neuroinflammation. Interestingly, the combined expression of TDP-43 and Aβ42 produced pathology similar to TDP-43 alone but overt neuronal loss occurred, indicating a possible synergistic effect between the two proteins. Another study using lentiviruses to express TDP-43 in the motor cortex found altered amino acid metabolism, oxidative stress, and neuronal death [135].
The studies described above demonstrate the versatility and utility of using rAAV and lentiviruses to produce models of neurodegenerative diseases. These tools can be used alone or in conjunction with existing transgenic models to help address potential pitfalls of previous modeling techniques. These valuable techniques will continue to provide insight into neurodegenerative diseases by allowing elucidation of mechanisms and providing platforms to explore new therapeutics.
11 Possible Therapeutic Uses of Viral Vectors
Currently, there are no treatments that effectively slow or prevent AD and other tauopathies. While the precise mechanisms of degeneration are unknown, loss of cholinergic neurons in the basal forebrain and excitotoxicity are believed to contribute to the development of AD [136, 137]. Accordingly, cholinesterase inhibitors (donepezil, rivastigmine, tacrine, and galantamine), which reduce acetylcholine breakdown, are used to treat mild to moderate AD. In addition, a noncompetitive NMDA receptor antagonist (memantine) is prescribed for moderate to severe disease stages [138]. However, neither of these drugs slow or prevent disease progression and only provide temporary symptomatic improvement for some patients [139]. Thus, there is an urgent need to aggressively develop novel therapeutics for treating AD and other tauopathies.
Nerve growth factor (NGF) is an appealing treatment because it potently protects cholinergic neurons and improves the function of basal forebrain neurons [139, 140]. The delivery of NGF to the nucleus basalis of Meynert was approached by direct infusion into the brain , implantation of cell grafts of NGF-secreting fibroblasts, and viral-vector delivery using adeno-associated virus . NGF injection into the lateral cerebral ventricle led to pain syndrome and weight loss with no obvious cognitive improvement in patients [141]. The delivery of NGF via fibroblast provided some therapeutic benefits in nonhuman primates, but the complexity of manufacturing fibroblasts and the diminished protein expression after 18 months makes it less desirable [142]. Using a rAAV vector to deliver NGF to the target region of the forebrain eliminates the hurdles of other NGF delivery methods. A phase I clinical trial (NCT00087789) was conducted using CERE-110 for the treatment of AD patients. The CERE-110 viral vector is an AAV2/2 vector containing full length NGF transgene under the control of the CMV/β-actin promoter and the human polyadenylation signal [143]. There were few adverse effects of the treatment reported, and all were deemed either not related or unlikely related to the rAAV-NGF treatment [139]. The phase I trial confirmed that viral vector therapy was well tolerated with a high level of safety and no systemic toxicity. The success of the phase I trial led to a phase II clinical trial using CERE-110 and results are expected in 2015. As our knowledge of disease mechanisms continues to grow and novel therapies are developed, gene therapy must continue to be pursued as a viable approach for therapeutic interventions in AD and other tauopathies.
Acknowledgements
This work was supported by NIH/NIA grant R01AG044372 (N.M.K.).
References
1.
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82(4):239–259PubMed
2.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82(12):4245–4249PubMedCentralPubMed
3.
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease [see comments]. Nature 349(6311):704–706PubMed
4.
Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T et al (1995) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376(6543):775–778. doi:10.1038/376775a0 PubMed
5.
Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H (1999) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A 96(2):742–747PubMedCentralPubMed
6.
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156(1):15–20PubMedCentralPubMed
7.
Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT (1992) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42(3 Pt 1):631–639PubMed
8.
Duering M, Grimm MO, Grimm HS, Schroder J, Hartmann T (2005) Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiol Aging 26(6):785–788. doi:10.1016/j.neurobiolaging.2004.08.002 PubMed
9.
Deshpande A, Mina E, Glabe C, Busciglio J (2006) Different conformations of amyloid beta induce neurotoxicity by distinct mechanisms in human cortical neurons. J Neurosci 26(22):6011–6018. doi:10.1523/JNEUROSCI.1189-06.2006 PubMed
10.
Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL (2007) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27(11):2866–2875. doi:10.1523/JNEUROSCI.4970-06.2007 PubMed
11.
Marin N, Romero B, Bosch-Morell F, Llansola M, Felipo V, Roma J, Romero FJ (2000) Beta-amyloid-induced activation of caspase-3 in primary cultures of rat neurons. Mech Ageing Dev 119(1–2):63–67PubMed
Related posts:
 Lentivirus Production and Purification
Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection
Gene Therapy for the Treatment of Neurological Disorders: Amyotrophic Lateral Sclerosis
Lentivirus Production and Purification
Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection
Gene Therapy for the Treatment of Neurological Disorders: Amyotrophic Lateral Sclerosis
 Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors
Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors
 Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
Gene Therapy of the Peripheral Nervous System: Celiac Ganglia
 Convection Enhanced Delivery of Recombinant Adeno-associated Virus into the Mouse Brain
Convection Enhanced Delivery of Recombinant Adeno-associated Virus into the Mouse Brain
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


