Yushi Inoue
Kiyohito Terada
Olivier Dulac
Guido Rubboli
Carlo Alberto Tassinari
Signa epilepsiae remotoria sunt jactatio et cetera. (Heberden, 1804)
Introduction
“The term and concept of myoclonus have been a longstanding source of confusion and debate. Myoclonus should be used in its strictly etymological sense, referring to any brief muscular contraction, as would have been wished by Muskens, the first neurologist to do detailed clinical and experimental studies of it.”42
The development of the concept of myoclonus and its application to epileptic seizures is closely connected with the history of description and conceptualization of progressive myoclonus epilepsy (PME) and other myoclonic epilepsies. Dubini (1846)35 described a cohort of patients with possibly different conditions who had involuntary jerky movements that he named electric chorea. Delasiauve (1854), Reynolds (1861), Féré (1890), Binswanger (1899), and Gowers (1902) described sudden starts or jerks as expressions of motor prodromes, auras, or abortive seizures (petit mal moteur) in their monographs on epilepsy. They did not, however, differentiate them semiologically or nosologically. Credit for the first exact and comparable description of these phenomena belongs to Herpin (1867),59 who described them as: “this variety of [seizure] prelude is a jerk [secousse] which runs through the entire body like an electric current.” His twenty-fourth observation was the first description of juvenile myoclonic epilepsy (JME), in a 14-year-old child, the son of a doctor. His jerks were called commotions or impulsions.
“At the beginning of the seizure, the movements or jerks were limited to the upper part of his body; they later became generalized. If the boy were to stand or walk, he might fall, although this is rare. He stands up again immediately [after the seizure]. He throws or drops what he is holding, particularly if he is holding it with his right hand. He says that he cannot see at the moment of the movement, but that he recovers immediately afterwards. His mother called these events (accidents) shakes (tremblement) and the father jerks (secousses).”
In 1881, Friedreich40 described a case, the diagnosis of which has remained unclear until today, and named it paramyoclonus multiplex. Many publications followed his observation, but they were comparable only in that they were dealing with involuntary jerks. From the “chaos of motor neuroses and from the cloudy pool of many publications,” Unverricht116 crystallized a unique disease entity from his 1895 study of members of two families and named it familiäre myoclonie. This disease started between the ages of 7 and 15 years, and consisted of a combination of nocturnal convulsive seizures and “lightning-like, arrhythmic, isolated jerks of individual, functionally unrelated muscles of both sides of the body. The jerks occurred less frequently when strained, but more frequently during psychic excitement, and stopped completely during sleep.” Unverricht seemed to have not observed any relationship between the phenomena he named myoclonie and the preparoxysmal or interval jerks of epileptic patients that, at that time, were still called secousses.
In 1899, both Dide27 and Rabot,90 at almost the same time, introduced the concept of myoclonie to the vocabulary of epilepsy—Dide from a semiologic viewpoint (“La myoclonie dans l’épilepsie”) and Rabot from a nosologic view (“De la myoclonie épileptique”). Rabot confirmed Herpin’s observation and emphasized that the “jerks” might precede major convulsive seizures for many years, usually occur in the morning after awakening, are prone to recur in series, and occur more frequently days before major convulsive seizures and disappear thereafter for a while, as if switched off.
Some years later, Clark and Prout19 proposed the concept of myoclonus epilepsy for their cases of Unverricht myoclonie, which they had described as paramyoclonus multiplex associated with epilepsy in 1900. When, in 1903, Lundborg76 sought an appropriate term for the disease he observed in 18 members of a Swedish farm family, he noticed the same nomenclature for two different diseases. He then proposed to describe one, which was “identical with the Unverricht’s myoclonie,” as progressive familial myoclonus epilepsy—from which later the term Unverricht-Lundborg myoclonus epilepsy developed—and the other, which was “standing near the essential epilepsy,” as intermittent sporadic myoclonus epilepsy (Rabot’s type). This was described by Lecasble in 1958 as “épilepsie generaliseé avec des myoclonies intermittentes et sporadiques” and by Castells and Mendilaharsu18 in the same year as “la epilepsia mióelonia bilaterally cosciente” to differentiate it from the epilepsy beginning with partial myoclonus. Janz66,67 as well as Lennox73 tried to replace the vague but accepted concept of myoclonus with the descriptive terms—at least for the jerks of JME—impulsive-petit mal67 (from the Latin impulsio, “jerk”) and jerk epilepsy.73 During the 1970s, a syndromic approach to the problems of myoclonic epilepsies occurred, with a new importance given to electroencephalographic (EEG) findings and to elements of etiology and prognosis. The Marseille school, among others, established several well-accepted syndromic entities using the adjective myoclonic, such as benign and severe myoclonic epilepsy in infancy, and epilepsy with myoclonic absences.44
Recently, myoclonus has been the subject of intensive research using advanced technologies. Electrophysiologic studies, especially, have brought new insights into the pathophysiomechanism of myoclonus.102 The recent advances in molecular genetics have also helped achieve an understanding of the different conditions that cause myoclonus.
Clinical Manifestations
Myoclonus consists of involuntary, quick movements distinctive for their abrupt, lightning-like character, occurring in a mild or vehement manner, spontaneously or in stimulus-sensitive conditions, affecting the entire body or in regional or localized distal or proximal parts of the body. It may be diffuse, predominating symmetrically in the axial muscles and the upper limb (massive myoclonus), or segmental, affecting randomly the different parts of the body, although the most involved muscles are those of the extremities, face, and trunk (erratic myoclonus); if the legs are involved, the person may be thrown to the ground. Myoclonus can be asymmetrical or even focal, limited to a restricted part of one limb or the face (epilepsia partialis continua).36 It may occur at random over time, or at more or less fixed interval, and in sporadic or rhythmic fashion. Most myoclonia are caused by abrupt muscle contraction (positive myoclonus); rarely, a brief interruption of ongoing activity may be observed (negative myoclonus, previously named asterixis).1 No distinguishable, even momentary, loss of consciousness occurs with single jerks, but if jerks occur in rapid succession or in the form of myoclonic status epilepticus, there may be a blurring of awareness.
Phenomenologically, epileptic myoclonus cannot be distinguished from the brisk, nonepileptic movements associated with physiologic or pathologic conditions. The classification of seizures as an epileptic manifestation requires knowledge of the clinical context and/or electrophysiologic confirmation, although physiologic correlates are not always demonstrable. Hallett54 proposed to define epileptic myoclonus as an elementary electroclinical manifestation of epilepsy involving descending neurons, whose spatial or temporal amplification can trigger overt epileptic activity.
In this chapter, we use the term myoclonic seizure as synonymous with epileptic myoclonus. The term myoclonus was coined by Friedreich.55 Myos means muscles, clonus means tumult or a quick movement, and (myo)clonia is the plural. Because a myoclonic movement is a jerk, the term myoclonic jerk is redundant.42
Stimulus Sensitivity
Myoclonus is often stimulus-sensitive, being elicited by stimuli of a single or multiple modalities. Tendon tap, posturing, passive or voluntary movement, visual and acoustic stimuli, and cognitive and emotional experiences may precipitate myoclonus. Myoclonus can also be facilitated by various physiologic factors such as the sleep–wake cycle and fatigue.
Rhythmicity/Periodicity
Myoclonus usually occurs irregularly; however, it occasionally appears to be rhythmic. Bilateral, rhythmic, virtually continuous myoclonus can be observed in Angelman syndrome. Guerrini et al.46 suggested that small areas within the motor cortex are able to independently produce hypersynchronous, rhythmic neuronal discharges recruiting muscle activity similar to tremor. Cortical tremor, in the form of postural or action tremor, and showing the neurophysiologic characteristics of reflex cortical myoclonus, can be observed in patients with PME and with benign adult familial myoclonic epilepsy (BAFME).60,111
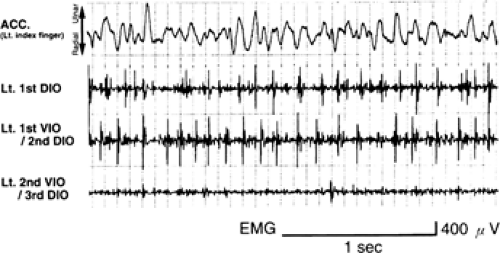 FIGURE 1. Polygraphic EMG records and accelerogram (ACC) in a patient with cortical myoclonus, obtained while the patient maintained the posture of finger extension. Each EMG activity lasted less than 50 msec. EMG discharges are relatively rhythmic. The ACC, therefore, demonstrates relatively sinusoidal wave corresponding to so-called “cortical tremor.” From Terada K, Ikeda A, Mima T, et al. Familial cortical myoclonic tremor as a unique form of cortical reflex myoclonus. Mov Disord. 1997;12:370–377, with permission. |
Epilepsia partialis continua is another example of the rhythmic, periodic, or arrhythmic recurrence of myoclonus. The cortical site of origin of the spike discharges and the site of origin of the associated focal myoclonic seizures do not necessarily coincide.22
Pathophysiology
The term epileptic myoclonus implies a central nervous system origin, presumably accompanied by neuronal depolarization shifts with subsequent hyperpolarizing potentials. To clarify the underlying pathophysiologic mechanisms of myoclonus, electrophysiologic studies are useful (i.e., electromyogram [EMG], somatosensory evoked potential [SEP], C-reflex, and jerk-locked back averaging [JLA]). Hallett et al.54,57 divided epileptic myoclonus into three categories: cortical reflex myoclonus, primary generalized epileptic myoclonus, and reticular reflex myoclonus. Guerrini et al.48 also divided this disorder into cortical, thalamocortical, and reticular form.
From an EMG point of view, myoclonus appears as short bursts of synchronized activity, which may involve agonist and antagonist muscles at the same time. The duration of EMG burst during epileptic myoclonus ranges between 10 and 100 msec, usually less than 50 msec (Fig. 1), and that of the EMG silent period of epileptic negative myoclonus from 50 to 400 msec (Fig. 2).44 By recording the EMG, the nature of myoclonus can be described (i.e., positive or negative myoclonus, duration of EMG burst, rhythmicity and periodicity, synchrony of agonist and antagonist muscles, distribution of myoclonus, spreading pattern of myoclonus, and so on).
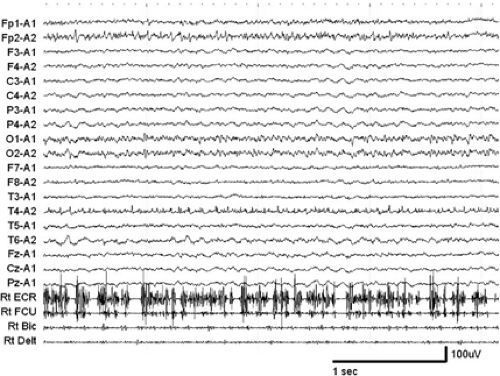 FIGURE 2. Polygraphic EEG/EMG records in a patient with ENM. The record was obtained while the patient maintained an extended posture of right wrist. SPs are seen in the right wrist extensor (ECR) and flexor (FCU) muscles synchronously. They are sometimes preceded by positive myoclonus. EEG shows no clear spikes in association with the jerks in this polygraphic recording. |
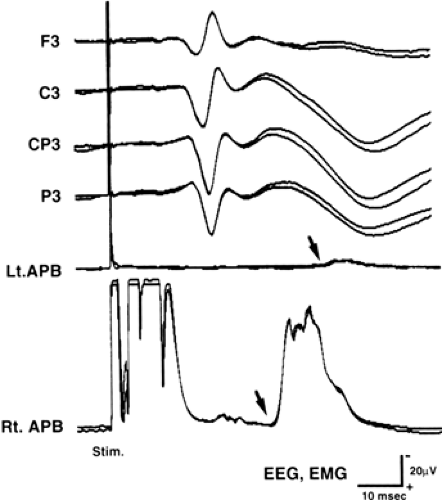 FIGURE 3. SEP and C-reflex following electric stimulation of the right median nerve in a patient with cortical myoclonus. The amplitudes of all SEP components except for N20 are abnormally large. Enhanced C-reflex is seen not only on the stimulated but also on the contralateral side. The arrows indicate the onset of C-reflex in abductor pollicis brevis (APB) muscles on each side. FromTerada K, Ikeda A, Mima T, et al. Familial cortical myoclonic tremor as a unique form of cortical reflex myoclonus. Mov Disord. 1997;12:370–377, with permission. |
Cortical Myoclonus
Cortical myoclonus reflects impulses that originate in the sensorimotor cortex and travel down the brainstem. Cortical myoclonus is typically seen in progressive myoclonus epilepsy. Muscles involved tend to be distal more than proximal and flexor more than extensor, and to involve more the face and upper extremities than the rest of the body. Cortical myoclonus is more commonly encountered in a multifocal form, presenting with multifocal spike discharges. If myoclonus is triggered by stimuli, the term cortical reflex myoclonus is used. If myoclonus occurs periodically, the term epilepsia partialis continua is used. The neurons in the sensorimotor cortex may be primarily hyperexcitable, or may be driven by abnormal inputs from the neurons of other brain parts. Therefore, cortical myoclonus occasionally is called fragmented epileptic convulsion.
In patients with cortical reflex myoclonus, the cortical components of median-nerve SEP showed abnormally large amplitude (Fig. 3). Usually, the initial peaks (N20/P22) are not large, and the following components become higher. This giant SEP is thought to indicate hyperexcitability of the sensorimotor cortex. Abnormally large evoked potentials were also reported by photic stimulation.104
When the peripheral nerve is stimulated, the stimulus goes up the spino-thalamo-cortical tract and, after excitation of the pyramidal neuron, it goes down the cortico-spinal tract, resulting in muscle contraction (long-loop reflex). In normal subjects, long-loop reflex can be recorded only when subjects maintain muscle contractions. In patients with cortical reflex myoclonus, however, this reflex can be recorded even while resting (C-reflex) (see Fig. 3). The latency of C-reflex for median nerve stimulation is about 40 to 45 msec, which is almost double of the latency of N20 to the median nerve stimulation. When the C-reflex is recorded from the contralateral limbs to the stimuli, the latency delay is about 10 msec to the ipsilateral limbs, which corresponds to the traveling time of the transcallosal pathway. This stimulation-locked muscle contraction is believed to share the same underlying mechanism with cortical reflex myoclonus.
Some EEG correlates are time-locked to cortical myoclonus. However, because of the relatively smaller amplitude of the EEG spikes in comparison with the background activities, the physiologic correlates of myoclonus can only be detected by using jerk-locked (EEG or magnetoencephalograhic [MEG]) averaging (JLA of jerk-locked magentic field [JLF]) (Fig. 4) or coherence analysis method.46,102 In JLA, EEGs are averaged with respect to the EMG onset, to reduce the non–time locked background EEG activities. Positive peak of the EEG spikes is 15 to 20 msec prior to the myoclonus for the upper limbs, and 25 to 40 msec for the lower limbs. Spikes are located around the contralateral primary motor cortex.
As such, cortical reflex myoclonus is caused by hyperexcitability of the primary sensorimotor cortex. However, because giant SEPs are not always present in patients with cortical reflex myoclonus (as in dentatorubral-pallidoluysian atrophy [DRPLA]), some other pathophysiologic mechanisms may exist.
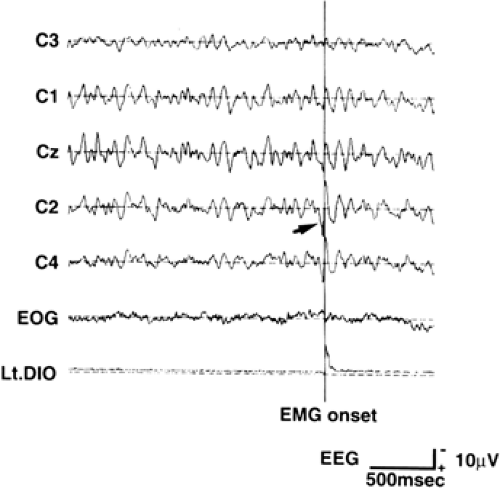 FIGURE 4. Waveforms of cortical discharge demonstrated by jerk-locked back averaging time-locked to the involuntary EMG activity of the left dorsal interosseus muscle (DIO) in a patient with cortical myoclonus. The positive/negative biphasic spike (arrow) is maximal at the right central area (C2), which precedes the EMG onset by about 30 msec. From Terada K, Ikeda A, Mima T, et al. Familial cortical myoclonic tremor as a unique form of cortical reflex myoclonus. Mov Disord. 1997;12: 370–377, with permission. |
In Lennox-Gastaut syndrome (LGS), myoclonus is rare and disclosed only in those cases with a cortical lesion affecting the rolandic area;12 thus, myoclonus appears to be produced by a secondary generalization of focal cortical myoclonus. They also present with arrhythmic, distal small focal jerks, leading to the individual tiny finger movements unaccompanied by premyoclonic potentials on JLA that Wilkins et al.119
proposed to call minipolymyoclonus. Brown et al.14 indicated that the major role of facilitation of inter- and intrahemis-pheric spread of cortical myoclonic activity is through trans-callosal or intrahemispheric corticocortical pathways in producing generalized or bilateral myoclonus. Therefore, bilateral jerks may not be synchronous in patients with cortical myoclonus.
proposed to call minipolymyoclonus. Brown et al.14 indicated that the major role of facilitation of inter- and intrahemis-pheric spread of cortical myoclonic activity is through trans-callosal or intrahemispheric corticocortical pathways in producing generalized or bilateral myoclonus. Therefore, bilateral jerks may not be synchronous in patients with cortical myoclonus.
Thalamocortical Myoclonus
Thalamocortical myoclonus or idiopathic generalized epileptic myoclonus represents the common type of myoclonus in various epileptic syndromes. Myoclonia are often spontaneous, predominantly arrhythmic and axial with varying severity, and associated chronologically with an EEG pattern of diffuse polyspikes or (poly) spike-and-wave discharges. A hyperexcitable cortex is thought to be driven diffusely and synchronously by ascending subcortical inputs that trigger the paroxysmal events. As a consequence, muscles from both sides are activated, and muscles innervated by the cranial nerves are involved through a rostrocaudal manner.
Electrophysiologically, SEP usually does not show giant SEP, and C-reflex may be recorded at rest. A negative peak of the generalized spike (30–100 msec duration) precedes the jerk (<100 msec duration) by 20 to 75 msec. The latency of the spike is relatively longer, and the temporal relationship is looser than in that of cortical myoclonus. The underlying mechanism of the thalamocortical myoclonus is still uncertain. The myoclonus of benign myoclonic epilepsy of infancy, myoclonic-astatic epilepsy, and JME belongs to this category.46
Myoclonus observed in patients with Dravet syndrome is not straightforward: Patients may exhibit massive myoclonus combined with a generalized spike-wave (rarely in infancy, mostly in childhood),32 and erratic myoclonus, particularly during episodes of myoclonic status, in which the patient is drowsy with diffuse slow wave activity and few spikes. The generator remains unidentified.
Reticular Reflex Myoclonus
Reticular reflex myoclonus originates in a hyperexcitable caudal brainstem reticular formation, giving rise to a widespread pattern of muscle activation with proximal and flexor predominance, spontaneous or induced by various stimuli. The impulses may travel up the brainstem. Reticular reflex myoclonus may be present simultaneously with cortical myoclonus.
Reticular reflex myoclonus is not time-locked to EEG discharges, and the sensory evoked potentials are not enhanced. Myoclonus is triggered by stimuli, but the temporal relationship is variable between the stimuli and the myoclonus, whereas it is constant in patients with cortical reflex myoclonus. The EMG discharges start in the areas of lower cranial nerves (sternocleidomastoid muscle, trapezius muscle). They go up to the facial muscles, down to the upper limbs, then to lower limbs. Therefore, it was speculated that the stimuli excited the reticular formation and that abnormal electrical activity then spread from it to the upper brainstem and the spinal cord.56
Other Myoclonus
Epilepsy generally does not occur with these other conditions. In spinal myoclonus, the jerks are generated in the spinal cord. Propriospinal myoclonus, nocturnal myoclonus, and periodic leg movements may be involved in this category.
Clinical and Electrographic Features of Myoclonic Seizures
Patients may refer to their myoclonus as a jerk, shock, jump, start, involuntary movement, or even stiffness and numbness. Patients may describe being thrown down, as though pushed by someone. Usually, patients do not notice any warnings, but sometimes smaller seizures serve as precursors to more severe ones. Myoclonus may be predominant in axial muscle groups or in the distal ones. Injury caused by falls or by striking a nearby object with a hand or limb may occur during an attack. Myoclonus occurs spontaneously, sometimes predominantly on awakening, and may be precipitated by various internal or external, simple or complex stimuli. Myoclonus may occur in isolation or in patients experiencing generalized tonic–clonic seizures (GTCs) or other forms of epileptic attacks.
In the following section, we describe the clinical and electrographic features of myoclonus in generalized forms. Because the clinical characteristics and EEG expressions of myoclonus are distinct in accordance with the epileptic syndromes, the descriptions accompany the syndromic classification. Detailed descriptions of the individual syndromes can be found elsewhere.92
Myoclonic Epilepsies of Infancy and Early Childhood
Harper58 called attention to the fact that myoclonic seizures occur in childhood, just as in adolescence or adulthood; he described true myoclonic epilepsy in childhood in the age group of 3 to 7 years. Aicardi3,4 further argued that approximately two thirds of the myoclonic epilepsies occur during the first 5 years of life, and that the true myoclonic epilepsies of childhood—which should be delineated from epilepsies with other types of brief axial motor seizures, such as epileptic spasms and tonic seizures of the LGS—might be subdivided into several forms. International classification of epilepsies and epileptic syndromes21 covers several distinctive syndromes occurring in infancy and early childhood in which myoclonus is a main seizure type.
Early Myoclonic Encephalopathy
Clinical Features
Massive or axial bilateral myoclonus is the myoclonic feature of early myoclonic encephalopathy, a rare syndrome with several causes occurring during the first 28 days of life (see Chapter 224). In addition, erratic myoclonus frequently repeats as the earliest and main clinical events, with a tendency to shift incessantly from one part of the body to another in an anarchic and asynchronous way.5,7 Events may be restricted to a very small territory, such as a finger or eyebrow, but sometimes may involve a whole limb. Focal motor seizures and, in the later stage, epileptic spasms or tonic seizures also may appear. Affected infants have severe neurologic abnormalities along with therapy-resistant seizures, and more than half of them die by the age of 1 year.
Electrophysiologic Features
The EEG activity consists of complex bursts of asynchronous spikes and irregular, arrhythmic sharp waves and slow waves lasting 1 to 5 seconds and alternating with flat periods of 3 to 10 seconds (suppression-burst pattern). Normal background activity is absent. The paroxysmal bursts are sometimes synchronous with bursts of bilateral myoclonus. The EEG may later evolve toward atypical hypsarrhythmia.
Myoclonic Status in Nonprogressive Encephalopathies
Clinical Features
This condition is characterized essentially by the recurrence of atypical status combined with an impairment of contact and continuous jerks in infants suffering from a nonprogressive severe encephalopathy with profound cognitive deficit and hypotonia.23 The myoclonus is more or less rhythmic, asynchronous, and multifocal, affecting periorbital, perioral, and distal muscles. It may be followed by a brief silent period but, in some patients, a negative myoclonus may be predominant. This electroclinical picture is usually observed in children with Angelman syndrome and with 4p-syndrome, allowing an early diagnosis of these disorders.
Other seizure types are focal motor seizures, myoclonic absences, and rare generalized or unilateral clonic seizures. The recognition of this condition is important to exclude the assumption of progressive disease and to allow an adequate treatment against the worsening of the cognitive impairment.
Electrophysiologic Features
The EEG shows bursts of more or less diffuse, more or less synchronous, rhythmic or arrhythmic discharges of diffuse slow spike-waves, often fluctuating, during wakefulness and sleep. Between these bursts are inserted periods of variable duration without obvious paroxysmal discharges but with θ–wave activity of variable amplitude involving both central regions subcontinuously.23 Bilateral jerk may be time locked with a cortical spike.
Benign Myoclonic Epilepsy in Infancy
Clinical Features
Myoclonus in this rare syndrome occurs during the first or second years of life in normal children who often have a family history of seizures or epilepsy. The seizures are characterized by frequent, brief, mostly symmetrical myoclonus isolated or grouped in clusters and involving the axis of the body and limbs. These seizures provoke a head drop and an upward-outward movement of the upper limbs, with flexion of the lower limbs and sometimes a rolling of the eyeballs.30,31 They may repeat pseudorhythmically. They occur at any time of day but may be enhanced by drowsiness. In some patients, myoclonus can be triggered by photic stimulation, or by a sudden noise or contact. The myoclonus represents the only seizure type, except for occasional febrile convulsions and GTCs, that occasionally develops later during adolescence. Seizures do not occur in series, and flexion of the entire body, tonic seizures, or absence seizures are never observed. Myoclonus is easily controlled by appropriate medical treatment but psychomotor development may be delayed; hence, the term “benign” is disputed.
Electrophysiologic Features
On EEG, myoclonus is accompanied by a discharge of bilateral spike-wave or polyspike-wave (Fig. 5) that sometimes occurs in rapid succession at more than 3 Hz. The background activity is normal for the child’s age, and drowsiness and the early stage of sleep may activate bilateral spike-waves. Myoclonus of about 100 msec duration consists of symmetric, rostocaudal muscle activation, and a premyoclonus negative spike precedes a jerk by about 30 msec.
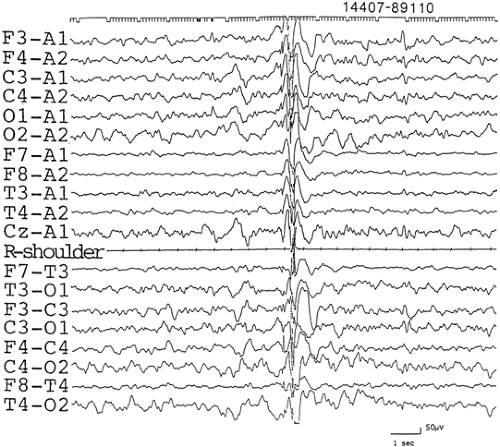 FIGURE 5. An EEG correlate of a myoclonus recorded in a boy, aged 2 years and 11 months, of benign myoclonic epilepsy in infancy. A brief burst of bisynchronous and symmetrical high-amplitude slow waves, maximal in the anterior head part and notched with small spikes, preceded the occurrence of muscle potentials of the right shoulder. The boy experienced daily axial myoclonus accompanied with abduction of the upper extremities starting at age 2 years and 8 months. |
Severe Myoclonic Epilepsy in Infancy (Dravet Syndrome)
Clinical Features
Dravet syndrome (see Chapter 230) is characterized by isolated or grouped axial-dominant bilateral myoclonus, which may lead to throwing of objects the child is holding and to falling. It is one of the typical features of severe myoclonic epilepsy in infancy. Symptoms appear between the ages of 2 and 5 years and occur very frequently, especially on awakening or in the hours preceding a major seizure. In the course of the disease, frequent, distally predominant and perioral erratic myoclonus that exists at rest but increases with movement may be seen, combined with drowsiness and drooling, in the context of nonconvulsive status epilepticus.
As a rule, this syndrome begins with a generalized or unilateral, often prolonged, febrile convulsive seizure before 1 year of age in normal infants.33,34 The seizures consist of myoclonic, clonic, or clonic–tonic–clonic seizures, atypical absences—often with clonic components—and complex
partial seizures. The seizures are extremely difficult to control, but myoclonus decreases and may disappear in the long-term course. Psychomotor development is retarded from the second year of life onward. This is accompanied by neurologic signs such as ataxia and slight pyramidal signs.
partial seizures. The seizures are extremely difficult to control, but myoclonus decreases and may disappear in the long-term course. Psychomotor development is retarded from the second year of life onward. This is accompanied by neurologic signs such as ataxia and slight pyramidal signs.
Myoclonus may be induced by variations in illumination, closure of the eyes, and fixation on patterns that can produce an autostimulation phenomenon.
Electrophysiologic Features
Myoclonus is accompanied by a burst of bisynchronous spike-waves or multiple spike-waves (Fig. 6), and appears to originate from spread of cortical myoclonus, whereas distal erratic myoclonus is often without EEG correlates, even on JLA.4,46 Photosensitivity, including pattern sensitivity, is rather common, and a photoconvulsive response may appear during the first 2 years of life—even as early as 4 or 5 months of age. The EEG background activity fluctuates, depending on the number and duration of the clinical seizure events. Paroxysmally, rhythmic θ-activity at 4 to 5 Hz appears in the centroparietal areas, as do fast bilateral spike-waves, which may be associated later with variable focal or multifocal abnormalities. This syndrome represents an epileptic encephalopathy with diffuse hyperexcitability of the brain, as revealed by sodium channel dysfunction.
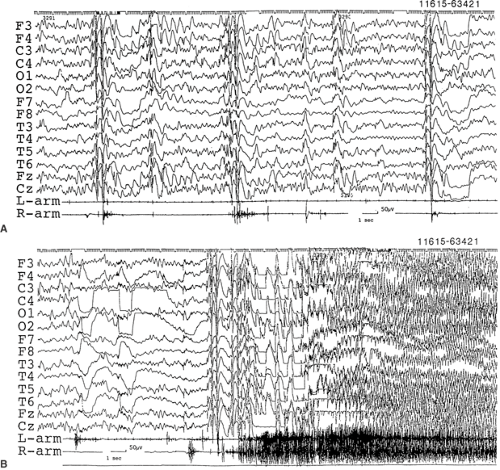 FIGURE 6. A: Frequent myoclonia shown as muscle potentials of arms concurrently occurring with bisynchronous and almost symmetrical polyspike-waves in EEG recorded in a 3-year-old boy. B: The myoclonus eventually cumulated into a generalized clonic–tonic–clonic seizure. He suffered from severe myoclonic epilepsy in infancy from 5 months of age on. Myoclonus became prominent from the age of 1 year and 2 months.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|


