Chapter 15 Genetic Basis of Sleep in Healthy Humans
Abstract
Evidence for Genotype-Dependent Differences in Diurnal Preference, Sleep Timing, Sleep Duration, Sleep Architecture, and Sleep EEG
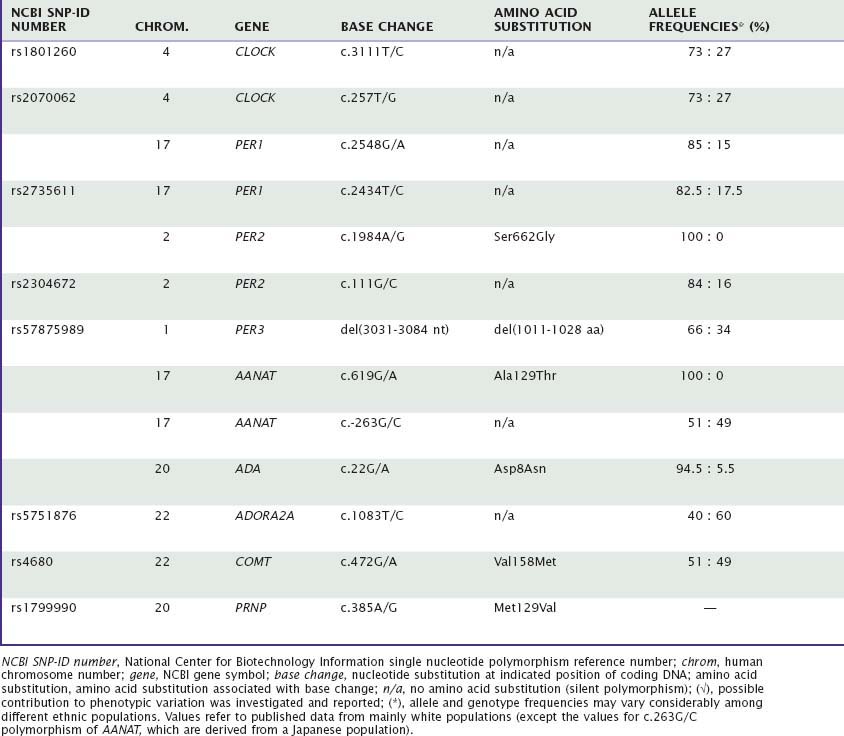
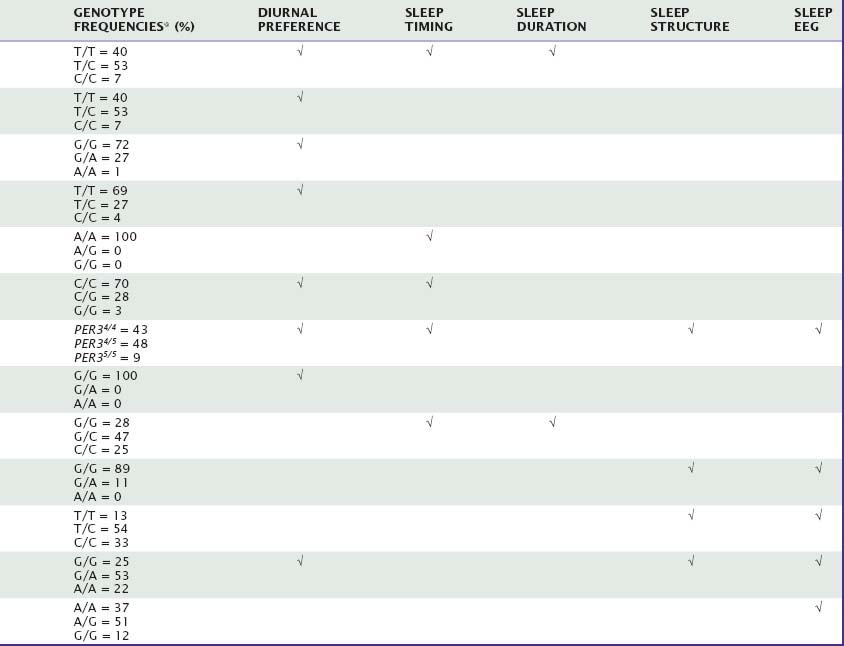
Many aspects of sleep and sleep–wake regulation are highly variable between individuals, yet highly stable within individuals. Uncovering genetic factors contributing to these traitlike individual differences in healthy humans constitutes one of the most promising avenues for fostering our understanding of the neurobiology of sleep in health and disease. This chapter will summarize the current evidence for genotype-dependent differences in duration, timing, and structure of sleep, as well as the sleep EEG, in healthy individuals. Table 15-1 summarizes the genetic variations in candidate genes that have been investigated to date to determine whether they contribute to genotype-dependent differences in diurnal preference, sleep timing, sleep duration, sleep structure, and sleep EEG. We will also review how these differences may relate to the homeostatic and circadian regulation of sleep. Several sleep characteristics differ between the sexes and between ethnic groups, but these differences will not be discussed here.
Two main techniques for the genetic dissection of normal human sleep are available. The first is to examine the impact of candidate genes—that is, genes for which evidence exists that they are involved in sleep and sleep–wake regulation. With this method, individuals with distinct genotypes of known genetic polymorphisms are prospectively studied in the sleep laboratory. This approach precludes discovery of novel “sleep genes,” but it may help us understand the consequences of these polymorphisms for sleep physiology. By contrast, genome-wide association studies may lead to the identification of novel “sleep genes,” which may lead to the discovery of novel sleep regulatory pathways. These studies, however, require very large sample sizes and multiple replications. The weaknesses and strengths of these strategies were recently discussed in detail.1
Large interindividual differences are observed in preferred time of day for completion of distinct cognitive tasks, sleep timing, sleep duration, sleep structure, and sleep EEG. Genes contribute to each of these phenotypes, and a high degree of heritability (i.e., the percentage of variance explained by overall genetic effects) has been demonstrated for these variables. For some of these variables, the magnitude of interindividual differences exceeds by far the size of the effects of manipulations of sleep regulatory processes, such as sleep deprivation.2
Genes Contributing to Human Morningness–Eveningness and Timing of Sleep
Candidate Genes
The timing of the peaks and troughs of daytime alertness and the timing of nocturnal sleep (i.e., diurnal preference) are highly variable among healthy individuals. Some of us go to sleep when others wake up. Self-rating scales such as the Horne-Östberg morningness–eveningness questionnaire (MEQ) and the diurnal type scale show normal distribution along a morningness–eveningness axis,3,4 indicating the contribution of additive, small effects of multiple genes in combination with the environment. Recent studies of large numbers of monozygotic (MZ) and dizygotic (DZ) twin pairs and of population- and family-based cohorts revealed roughly 50% heritability for diurnal preference5 and 22% to 25% for habitual bedtime.6,7
The effect of a single nucleotide polymorphism (SNP) in the 3′-untranslated region (UTR) of the human “Circadian locomotor output cycles kaput” gene (CLOCK), located on chromosome 4, on diurnal preference was first studied in middle-aged adults. This SNP may affect the stability and half-life of messenger RNA,8 and thus alter the protein level that is finally translated. Katzenberg and colleagues9 reported that homozygous carriers of the 3111C allele have increased evening preference for mental activities and sleep, with delays ranging from 10 to 44 minutes, when compared with individuals carrying the 3111T allele. A similar association with diurnal preference was found in a Japanese population, and MEQ scores were significantly correlated with sleep onset time and wake time.4 By contrast, studies in healthy European and Brazilian samples failed to confirm an association between genetic variation in CLOCK and diurnal preference.10,11 Interestingly, an almost complete linkage disequilibrium was shown between the 3111T→C and the 257T→G polymorphisms located in the other extremity of this gene.11 Full-length analysis of secondary mRNA structure revealed no interaction between the two polymorphisms.
Mouse Per1 and Per2 are importantly involved in maintaining circadian rhythmicity,12 and possible associations between variation in these genes and diurnal preference were thus also investigated in humans. Screening for missense mutations and functional or synonymous polymorphisms in promoter, 5′- and 3′-UTR, and coding regions of the period-1 gene (PER1) in volunteers with extreme diurnal preference and patients with delayed sleep-phase syndrome (DSPS) remained initially unsuccessful.13,14 By contrast, the distribution of the C and T alleles of a silent polymorphism in exon 18 was found to differ between extreme morning and evening types.14 Thus, the frequency of the 2434C allele was roughly double in subjects with extreme morning preference (24%) compared with subjects with extreme evening preference (12%). This polymorphism may be linked to another functional polymorphism, or it may directly affect PER1 expression at the translational level.14
A missense mutation in the human period-2 gene (PER2) currently provides the most striking example of a direct link with between genetic variation in a clock gene and changed circadian rhythms. Linkage analyses in families afflicted with familial advanced sleep-phase syndrome (FASPS) revealed associations with functional polymorphisms of PER2 that cause altered amino acid sequences in regions important for phosphorylation of this protein15 and a mutation in casein kinase delta (CKδ), which plays an important role in phosphorylation.16 The subsequent finding in a transgenic mouse model expressing the human FASPS mutation that casein kinase I delta (CKIδ) can regulate circadian period through PER2 provided further evidence that this gene is importantly involved in the mechanisms of circadian rhythm regulation in humans.17 In accordance with this notion, a C111G polymorphism located in the 5′-UTR of PER2 modulates diurnal preference in healthy volunteers.18 Thus, the 111G allele is significantly more prevalent in subjects with extreme morning preference (14%) than in individuals with extreme evening preference (3%). Computer simulation predicted that the 111G allele has a secondary RNA structure different from that of the 111C allele, and that the two transcripts may be differently translated.18
Findings in mice suggest that Per3 functions outside the core circadian clock work.12 Nevertheless, a variable-number tandem-repeat (VNTR) polymorphism in the human period-3 gene (PER3) also appears to modulate morning and evening preference. A 54-nucleotide sequence located in a coding region of this gene on human chromosome 1 is repeated in either four or five units. This difference may alter the dynamics in PER3 protein phosphorylation. The longer five-repeat allele was associated in European and Brazilian populations with morning preference, and the shorter four-repeat allele with evening preference, respectively.19,20
The gene encoding arylalkylamine N-acetyl-transferase (AANAT) is located on human chromosome 17q25. This enzyme plays a key role in melatonin synthesis and thus may be important for diurnal preference and circadian rhythm disturbances. Comparison in a Japanese population between 50 outpatients diagnosed with DSPS and 161 unrelated healthy controls suggested that the frequency of a seldom-occurring threonine allele at codon 129 is significantly higher in patients than in controls.21 This association was not confirmed in a Brazilian population, where virtually no allelic variation at this position was found.22 In a small study conducted in Singapore, it was suggested that a commonly occurring, silent 263G→C polymorphism of AANAT modulates sleep timing and sleep duration (also see later) in healthy students.23
Genome-Wide Association Study
Only one genome-wide association study of sleep-related phenotypes is currently available in humans.6 In the Framingham Heart Study 100K Project, phenotypic and genetic analyses were conducted in 749 subjects and revealed a heritability estimate for habitual bedtime of 22%. This small study suggests that a nonsynonymous polymorphism in a coding region of the gene encoding neuropeptide S receptor 1 (NPSR1) is a possible modulator of usual bedtime as obtained from a self-completion questionnaire. This polymorphism leads to a gain-of-function mutation in the receptor protein by increasing the sensitivity for neuropeptide S receptor 10-fold.24 Although a possible association of NPSR1 to weekday bedtime is interesting, the statistical power of this pilot study is limited, and the necessary replication of this finding in independent samples is lacking. A recent analysis of a larger sample of the Framingham Offspring Cohort did not parallel the prior result.7
Genes Contributing to Habitual Sleep Duration
Habitual sleep duration, like diurnal preference, shows large variation between healthy individuals, and the physiologic sleep and circadian correlates of habitual short and long sleepers have been identified in small groups of subjects.25–27 The temporal profiles of nocturnal melatonin and cortisol levels, body temperature, and sleepiness under constant environmental conditions and in the absence of sleep suggest that the circadian pacemaker programs a longer biological night in long sleepers than in short sleepers.27 Individual differences in this program may contribute to the large variation in habitual sleep duration, which shows a perfect normal distribution in the general population.28,29 Such a distribution is consistent with the influence of multiple, low-penetrance polymorphisms. Several older-twin studies and one recent genome-wide association study reported for sleep duration moderate heritability estimates of 17% to 40%.6,30–32
The Framingham Heart Study 100K Project revealed a linkage peak to usual sleep duration on chromosome 3 including the gene encoding prokineticin 2 (PROK2).6 This neuropeptide may be an important output molecule from the suprachiasmatic nucleus (SCN), in particular in defining the onset and maintenance of the circadian night.33,34 Because the danger of false-positive inferences from small genome-wide association studies is high, the methodological limitations of this work discussed earlier also apply to this potential association. It was not corroborated in a larger sample of the Framingham Cohort.7
Genes Contributing to Sleep Architecture
Many variables characterizing sleep architecture demonstrate large variation between individuals and high stability within individuals.2,35–37 For example, the intraclass correlation coefficients, which estimate the intraindividual stability of a variable across conditions (e.g., baseline versus sleep deprivation), was reported to be 0.73 for slow-wave sleep (SWS) and 0.48 for REM sleep.2 This observation suggests the presence of traitlike, interindividual differences in sleep physiology, which have a genetic basis. Indeed, twin studies show striking similarity and concordance in visually defined sleep variables in MZ twins, yet not in DZ twins. Already the first polysomnographic sleep studies in MZ twins have revealed almost complete concordance in the temporal sequence of sleep stages.38 Subsequent work showed that in particular those variables that most reliably reflect sleep need are under tight genetic control. Apart from total sleep time, they include duration of non-REM sleep stages, especially SWS, and density of rapid eye movements in REM sleep.39–41 Linkowski41 estimated that the heritability of REM density is up to 90%.
Slow-Wave Sleep and REM Sleep
A few studies have conducted polysomnographic assessment in defined genotypes. The CLOCK genotypes that were associated with diurnal preference9 did not significantly affect sleep variables derived from nocturnal polysomnography.
Only two polymorphisms have been identified to date as affecting sleep architecture in humans. First, in one study, homozygous carriers of the long-repeat genotype in PER3 (PER35/5), which associates with morningness, fell asleep more rapidly (about 9 versus 18 minutes) and showed more SWS (about 23% versus 16% of total sleep time) compared with homozygous four-repeat individuals.42 In addition, during recovery sleep from sleep deprivation, REM sleep was reduced in PER35/5 individuals.
Second, a functional variation in the gene on chromosome 20 encoding the enzyme adenosine deaminase (ADA) was found to have a profound impact on sleep architecture.43 Adenosine and adenosine receptors are thought to be involved in regulating distinct aspects of human sleep,44 and ADA plays an important role in regulating extracellular adenosine levels. Healthy individuals with the G/A genotype, associated with lower ADA activity in erythrocytes and leucocytes than in individuals with the G/G genotype, showed significantly more SWS in a baseline recording than subjects with the G/G genotype (about 93 versus 63 minutes). This difference is comparable to the difference between a baseline night and recovery night after one night without sleep. All other sleep variables were similar in both genotypes.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


