INTRODUCTION
Prevalence and Incidence
Alzheimer’s disease (AD) is the most common irreversible, progressive brain disease. It is characterized by a gradual loss of memory and cognitive skills. AD accounts for over 50% of all dementia cases and it presently affects more than 24 million people worldwide. Moreover, over 5 million new cases of AD are reported each year and the incidence is likely to increase as a greater proportion of the population ages1.
AD prevalence and incidence strongly suggest that age is the most influential known risk factor. Indeed, AD prevalence increases significantly with age and AD incidence increases from 2.8 per 1000 person years for people between 65 and 69 years to 56.1 per 1000 person years for people who are older than 90 years2. Approximately 10% of persons older than 70 years have significant memory loss and more than half of these individuals have probable AD. An estimated 25% to 45% of persons older than 85 years have dementia3. The duration of disease is typically 8 to 10 years, with a range from 2 to 25 years after diagnosis.
The disease is divided into two subtypes based on the age of onset: early-onset AD (EOAD) and late-onset AD (LOAD). EOAD accounts for approximately 1% to 6% of all cases and ranges roughly from 30 years to 60 or 65 years. On the other hand, LOAD, which is the most common form of AD, is defined as AD with an age at onset later than 60 or 65 years. Both EOAD and LOAD may occur in people with a positive family history of AD. Approximately 60% of EOAD cases have multiple cases of AD within their families and of these familial EOAD cases, 13% are inherited in an autosomal dominant manner with at least three generations affected4,5. Early- onset cases can also occur in families with late-onset disease3. With the exception of a few autosomal dominant families that are singlegene disorders (see below), most AD cases appear to be a complex disorder that is likely to involve multiple susceptibility genes and environmental factors3,6-9.
Clinical Symptoms
Both EOAD and LOAD present clinically as dementia that begins with a gradual decline of memory and then slowly increases in severity until the symptoms eventually become incapacitating. Other common symptoms are confusion, poor judgement, language disturbance, agitation, withdrawal and hallucinations. Rare symptoms include seizures, parkinsonism, increased muscle tone, myoclonus, incontinence and mutism. Death commonly occurs from general inanition, malnutrition and pneumonia3. Treatment of AD with cholinesterase inhibitors and memantine may result in slowing of cognitive decline in mild to moderate dementia cases, but overall, most patients experience only clinically marginal improvement in measures of cognition and in global assessments of dementia10,11.
Clinical Diagnosis
Currently, the diagnosis of AD is based on clinical history and neuropsychological tests. The Diagnostic and Statistical Manual of Mental Disorders, 4th Edition (DSM-IV), criteria for diagnosing dementia requires the loss of two or more of the following: memory, language, calculation, orientation or judgement12. The Mini-Mental State Examination (MMSE) helps to evaluate changes in a patient’s cognitive abilities. In addition, a diagnosis of probable AD necessitates the exclusion of other degenerative disorders associated with dementia, such as frontotemporal dementia (including frontotempo- ral dementia with parkinsonism-17 and Pick’s disease), Parkinson’s disease, diffuse Lewy body disease, Creutzfeldt-Jakob disease and cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL)13. Discriminating AD from other forms of dementia is usually done through clinical history and neuroimaging3. In addition, other possible causes of dementia need to be excluded, especially the treatable forms of cognitive impairment, such as impairment due to depression, chronic drug intoxication, chronic central nervous system infection, thyroid disease, vitamin deficiencies (e.g., B12 and thiamine), central nervous system angitis and normal-pressure hydrocephalus3. Individuals who do not meet these criteria but have short-term memory loss, have only minimal impairment in other cognitive abilities and are not functionally impaired at work or at home are considered to have ‘mild cognitive impairment’14.
Neuropathological Diagnosis
A definitive diagnosis of AD requires a clinical assessment of probable AD, as well as postmortem confirmation, with the presence of two histopathological features: neurofibrillary tangles and amyloid plaques15-17. Before autopsy confirmation, expert clinicians correctly diagnose AD between 80% to 90% of the time18. Even though plaques and tangles are often found in cognitively normal age- matched controls, the density and the distribution of these attributes are more severe in patients with AD, according to standardized histological assessments15. Amyloid plaques are extracellular with a cross-beta structure that is characteristic of dye-binding (neuritic amyloid plaques contain thioflavin S and Congo red-positive fibrillar deposits with both Aβ40 and Aβ42 present)19,20. The major component of amyloid plaques is amyloid beta (Aβ), which can be stained and detected using Aβ antibodies21,22. The most common form of Aβ in humans is 40-amino-acids long and is called Aβ40. A 42-amino-acid-long fragment, Aβ42, is less abundant than Aβ40 and differs only in that it has two additional amino-acid residues at the C-terminus. Aβ42 is associated with AD23.
Aβ is derived from the amyloid precursor protein (APP) by the action of two aspartyl proteases. First, α-secretase (non-neurotoxic ‘normal’ cleavage) or β-secretase (potential neurotoxic ‘abnormal’ cleavage) cleave APP (Figure 43.1) and second, γ-secretases cleave APP24 – 26. Upon cleavage by α-secretase, APP releases a large soluble APP alpha fragment (sAPPa) and a membrane-bound C-terminal 83-amino-acid fragment (C83); whereas upon cleavage by β-secretase, APP releases a large soluble APP beta fragment (sAPPβ) and a membrane-bound C-terminal 99-amino-acid fragment (C99)27,28. The C99 fragment is subsequently cleaved by γ-secretase to release Aβ and an APP intracellular domain (AICD)29,30 (Figure 43.1). Thus, depending on the point of cleavage
Figure 43.1 APP cleavage. The APP protein can be cleaved by three different secretases; α, β or γ (panel A). Subsequent to ‘normal’ α-secretase cleavage, sAPPα is produced and released into the extracellular space and the C83 peptide remains in the cell membrane (panel B). Subsequent to β-secretase cleavage, sAPPβ is produced and released into the extracellular space and the C99 peptide remains in the cell membrane (panel C). Subsequent to β-secretase cleavage, the C99 peptide is ‘abnormally’ cleaved by γ-secretase to yield an Aβ peptide and the AICD peptide (panel D). Scale is approximate
Source: Reproduced from Bekris, Yu, Bird & Tsuang (2010). The Genetics of Alzheimer’s and Parkinson’s Disease. In: A. Lajtha (Ed.), Handbook of Neurochemistry and Molecular Neurobiology, 3rd edn. Neurochemical Mechanisms in Disease, Vol. 36. New York: SpringerVerlag. With kind permission of Springer Science+Business Media
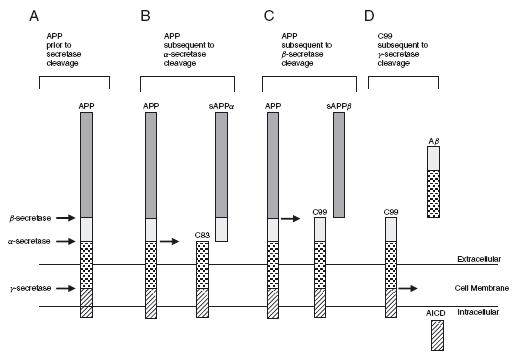
by γ-secretase, two main forms of Aβ are produced, consisting of either 40 or 42-amino-acid residues (Aβ40 or Aβ42). The proportion of Aβ40 to Aβ42 that is formed is particularly important in AD because Aβ42 is far more prone to oligomerize and form fibrils than the more abundantly produced Aβ40 peptide. Indeed, although it appears that the production of Aβ isoforms is a normal process of unknown function, in a small number of individuals, an increased proportion of Aβ42 appears sufficient to cause EOAD16,31.
Neurons bearing neurofibrillary tangles are another frequent finding in AD brains32,33 and the temporal and spatial appearance of these tangles, which contain hyperphosphorylated tau, more closely reflects disease severity than does the presence of amyloid plaques34,35. Tangles are formed by hyperphosphorylation of a microtubule- associated protein known as tau, causing it to aggregate in an insoluble form. However, neurofibrillary tangles are also found in other disorders, such as frontotemporal dementia and progressive supranuclear palsy. Moreover, these tangles are not necessarily associated with the cognitive dysfunction and memory impairment that is typical of AD and mutations in the gene that encodes the tau protein (MAPT), a main component of neurofibrillary tangles, has not been genetically linked to AD22.
GENETICS OF ALZHEIMER’S DISEASE
Introduction
Overall, more than 90% of AD cases appear to be sporadic and to have a later age at onset of 60 to 65 years (LOAD)36. Although twin studies support the existence of a genetic component in LOAD, no causative gene has been yet identified. Indeed, the only gene that has been consistently found to be associated with sporadic LOAD, across multiple genetic studies, is the apolipoprotein E (APOE) gene37-41 (Table 43.1). However, many carriers of the APOE risk allele (e4) live into their 90s, which suggests the existence of other LOAD genetic and/or environmental risk factors that have yet to be identified. To this end, several unreplicated genetic variants have been reported and these findings suggest that there may be five to seven major LOAD susceptibility genes3,42,43. For a catalogue of candidate gene association studies, please refer to the AlzGene online database (www.alzforum.org/res/com/gen/alzgene/default.asp).
Genes Associated with Autosomal Dominant Alzheimer’s Disease
AD1: Amyloid precursor protein (APP)
Inheritance and clinical features In the 1980s, Kang and colleagues purified both plaque and vascular amyloid deposits and isolated their 40-residue constituent peptide (Aβ), which subsequently led to the cloning of the APP type I integral membrane glycoprotein from which Aβ is proteolytically derived44. The APP gene was then mapped to chromosome 21q, which accounted for the observation that Down syndrome patients (trisomy 21) develop amyloid deposits and the neuropathological features of AD when in their 40s22,45 -47.
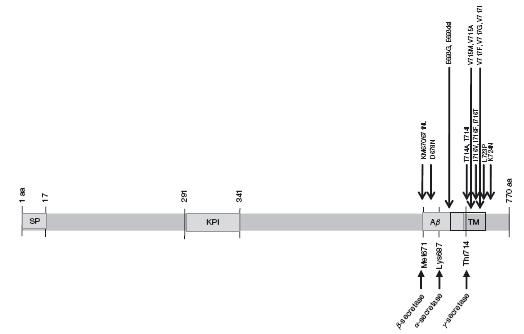
Since then, over 32 different APP missense mutations have been identified in 85 families. Interestingly, most of these mutations are located at the secretase cleavage sites or the APP transmembrane domain on exons 16 and 17 (Figure 43.2). Information regarding APP mutations are available in the NCBI database and the Alzheimer Disease Mutation Database (www.molgen.ua.ac.be/ADMutations)48. Mutations within APP account for 10% to 15% of early-onset familial AD (EOFAD)^49-51, appear to be family specific and do not occur within the majority of sporadic AD cases. The majority of these EOFAD mutations are in or adjacent to the Aβ peptide sequence (Figure 43.2), the major component of amyloid plaques52,53. Most cases containing APP mutations have an age of onset in the mid-40s and 50s54.
Gene location and structure Sequences encoding APP were first cloned by screening cDNA libraries44. The initial full-length cDNA clone encoded a 695-amino-acid protein (APP695)40 and consisted of 18 exons. The APP gene, located on chromosome 21q21, is alternatively spliced into several products, named according to their length in amino-acids (i.e., APP695, APP714, APP751, APP770 and APP563) and expressed differentially by tissue type whereby the three isoforms that are most relevant to AD are restricted to the central nervous system (APP695) or expressed in both the peripheral and CNS tissues (APP751 and APP770)44,55-61.
Gene function and expression APP is a type-I integralmembrane protein44 that resembles a signal-transduction receptor. It is expressed in many tissues and concentrated in the synapses of neurons. Its primary function is not known, though it has been implicated in neural plasticity62 and as a regulator of synapse formation63. APP is synthesized in the endoplasmic reticulum, post-transcriptionally modified in the golgi (N- and O-linked glycosylation, sulfation and phosphorylation) and transported to the cell surface via the secretory pathway. APP is also endocytosed from the cell surface and processed in the endosomal-lysosomal pathway64,65. APP and its byproduct Aβ have been found to be translocated inside mitochondria and implicated in mitochondrial dysfunction66-68.
Proteolysis of APP by α-secretase or β-secretase leads to the secretion of sAPPa or sAPPβ. This proteolysis generates C-terminal fragments of 10kDa and 12kDa, respectively, which are inserted into the membrane. These fragments can be cut by γ-secretase to
Table 43.1 Alzheimer’s disease (AD) genes. AD genes are located on four different chromosomes and are associated with autosomal dominant or sporadic inheritance
Source: Reproduced from Bekris, Yu, Bird & Tsuang (2010). The Genetics of Alzheimer’s and Parkinson’s Disease. In: A. Lajtha (Ed.), Handbook of Neurochemistry and Molecular Neurobiology, 3rd edn. Neurochemical Mechanisms in Disease, Vol. 36. New York: Springer-Verlag, Table 1. With kind permission of Springer Science+Business Media
Figure 43.2 AD1: APP structure and mutations. SP: Signal peptide. KPI: Kunitz protease inhibitor domain. Aβ : Amyloid beta. TM: Transmembrane domain. Scale is approximate
Source: Reproduced from Bekris, Yu, Bird & Tsuang (2010). The Genetics of Alzheimer’s and Parkinson’s Disease. In: A. Lajtha (Ed.), Handbook of Neurochemistry and Molecular Neurobiology, 3rd edn. Neurochemical Mechanisms in Disease, Vol. 36. New York: SpringerVerlag. With kind permission of Springer Science+Business Media
extracellularly release the Aβ peptide69 and intracellularly release a cytoplasmic fragment identified as AICD70 (Figure 43.1). The majority of EOFAD mutations alter this processing of APP in such a way that Aβ42 levels are changed relative to other Aβ isoform levels71,72. The function of these APP proteolytic fragments is still unclear.
The missense APP ‘Swedish’ mutations (APPSW, APPK670N, M671L) and the ‘London’ mutations (APPLON, APPV717I) are examples of APP mutations that lead to increased Aβ production and development of AD73,74. Transgenic mouse models of such APP mutations (PDAPP, Tg2576, APP23, TgCRND8 and J20) have been developed75. Each of these mouse models have different APP expression levels and different neurological abnormalities75,76. For example, the Tg2576 mouse model that carries the ‘Swedish’ mutation has high APP levels, high Aβ levels and cognitive disturbances77 that are progressive and start as early as six months of age78.
Genetic variation APP transcripts have been identified in which exons 7, 8 and 15 are alternatively spliced. APP695, the predominant isoform in neurons79, contains exon 15 but excludes exons 7 and 8. The APP751 isoform and the APP770 isoform both encode Kunitztype protease inhibitor (KPI)-containing forms of APPare present in both peripheral tissue and neurons and contain exon 16 and 17 but exclude exons 7, 8 and 15. The gene region that encodes the portion of APP that is cleaved to produce the Aβ peptide is located within exons 16 and 17 of the APP770 splice variant81 (Figure 43.2). Other splice variants have been observed that are missing exon 15 in various combinations with exons 7 and 8; these splice variants are referred to as L-APPs80,82. A number of studies have indicated that alternative splicing of exons 7 and 8 is modulated in brain during ageing and possibly during AD82-87. Even though the function of APP and its various splice variants is unknown, differential expression of these splice variants between tissues may imply functional differences. It is important to note that while most of the described splice variants contain Aβ-encoding sequences, two additional rare transcripts, APP365 and APP563 do not, implicating additional functionally important variability in APP isoform function55,88.
The first described and best characterized APP mutation (V717I) is located within the transmembrane domain near the γ -secretase cleavage site73 (Figure 43.2). Other substitutions have since been identified at this site and several groups have reported the V717I mutation in families who are unrelated to the initial London family. Many other APP mutations have since been identified, especially near the γ-secretase cleavage site, and the bulk of these mutations have been associated with modulation of Aβ levels. For example, a C-terminal L723P mutation was identified in an Australian family; this mutation is reported to generate an increase of Aβ42 peptide levels in CHO cells89. The majority of EOFAD mutations alter processing of APP in such a way that the relative level of Aβ42 is increased by increasing Aβ42, decreasing Aβ40 peptide levels or increasing Aβ42 while decreasing Aβ40 peptide levels71,72.
AD3: Presenilin 1 (PSEN1)
Inheritance and clinical features Linkage studies established the presence of an AD3 locus on chromosome 1490, and positional cloning led to the identification of mutations in the presenilin-1 (PSEN1 ) gene, which encodes a polytopic membrane protein91. Pre- senilins are major components of the atypical aspartyl protease complexes that are responsible for the γ-secretase cleavage of APP92,93. Mutations in PSEN1 are the most common cause of EOFAD. Indeed, PSEN1 missense mutations account for 18% to 50% of autosomal dominant EOFAD cases94. Over 176 different PSEN1 mutations have been identified in 390 families. PSEN1 mutations appear to increase the ratio of Aβ42 to Aβ40 and thereby result in a change in function that leads to reduced γ-secretase activity95. It has been suggested that deposition of Aβ42 may be an early preclinical event in PSEN1 mutation carriers96.
Defects in PSEN1 cause the most severe forms of AD, with complete penetrance and an onset occurring as early as 30 years of age. However, there is a wide variability in age of onset as other PSEN1 -associated AD cases have a mean age of onset greater than 58 years. PSEN1 -associated AD is an autosomal dominant neurodegenerative disorder characterized by progressive dementia and parkin- sonism, notch signalling modulation and Aβ intracellular domain generation16,97. There is also considerable phenotypic variability in PSEN1 -associated AD cases, including some patients who develop spastic paraparesis and other atypical AD symptoms. Some of these variable clinical phenotypes have been associated with specific mutations. Neuropathological studies often confirm the clinical diagnosis of AD with measurement of amyloid plaque and Braak stage (as described above) as well as other neurodegenerative changes in other brain areas according to the presence of specific PSEN1 mutations98,99. For example, researchers found that the clinical and neuropathological features of a Greek family with a PSEN1 mutation (N135S) included memory loss when the family members were in their 30s, as well as variable limb spasticity and seizures. Upon neu- ropathological examination of these family members, the diagnosis of AD was confirmed and there was also histological evidence of corti- cospinal tract degeneration99. Moreover, a PSEN1 mutation (I143M) that lies in a cluster in the second transmembrane domain of the protein has been described in an African family with an age at onset in the early 50s and a short duration of illness of six to seven years. These family members were diagnosed upon autopsy with severe AD pathology characterized by neuronal loss, abundant Aβ neuritic plaques and neurofibrillary tangles and degeneration extending into the brain stem100.
Gene location and structure PSEN1 is located on chromosome 14q24.2. It consists of 12 exons that encode a 467-amino-acid protein that is predicted to traverse the membrane 6 to 10 times; the amino and carboxyl termini are both oriented toward the cytoplasm101.
Gene function and expression PSEN1 is a polytopic membrane protein that forms the catalytic core of the γ -secretase complex92,102. Gamma-secretase is an integral membrane protein typically found at the cell surface, but it may also be found in the golgi, endoplasmic reticulum and mitochondria92,103.
PSEN1 , nicastrin (Nct), anterior pharynx defective 1 (Aph-1) and presenilin enhancer 2 (PSENEN) are required for the stability and activity of the γ-secretase complex104-108. This complex cleaves many type-I transmembrane proteins, including APP and Notch92,109, two proteins in the hydrophobic environment of the phospholipid bilayer of the membrane107.
PSEN1 knockout (KO) mice are not viable110 but a conditional PSEN1 KO mouse model, where the loss of the gene is limited to the postnatal forebrain, found that KO mice exhibited mild cognitive impairments in long-term spatial reference memory and retention111. These findings suggest that presenilins play a role in cognitive memory. Moreover, knockin mice with missense mutations of the endogenous murine PSEN1 have high Aβ42 levels and perform poorly on the object recognition test112,113. Double PSEN1 /APP transgenics have been developed and these transgenic suggest that PSEN1, APP and mutations within these genes play a role in the production of Aβ 76,114.
Genetic variation To date, there have been 176 PSEN1 mutations reported (Figure 43.3). A comprehensive list of PSEN1 mutations is available through the NCBI database (www.molgen.ua.ac.be/ADmutations). The majority of these mutations are missense mutations. These missense mutations cause amino-acid substitutions throughout the PSEN1 protein and appear to result in a relative increase in the ratio of Aβ42 to Aβ40 peptides; this increase seems to occur through increased Aβ42 production, decreased Aβ40 production, or alternatively, a combination of increased Aβ42 production and decreased Aβ40 production71. For example, individuals that carry the PSEN1 L166P mutation can have an age at onset in adolescence, and in vitro studies indicate that this mutation induces exceptionally high levels of Aβ42 production as well as impaired notch intracellular domain production and notch signalling98.
AD4: Presenilin 2 (PSEN2)
Inheritance and clinical features A candidate gene for the chromosome 1 AD4 locus was identified in 1995 in a Volga German AD kindred with a high homology to the AD3 locus (PSEN1 ); this gene was later named presenilin 2 (PSEN2 )51,115,116. In contrast to the mutations in the PSEN1 gene, missense mutations in the PSEN2 gene are a rare cause of EOFAD, at least in Caucasian populations. The clinical features of PSEN2 -affected families appear to differ from the clinical features of PSEN1 -affected families in that the age of onset in these family members is generally older (45-88 years) than for some family members with PSEN1 mutations (25-65 years). Furthermore, the age of onset is highly variable among PSEN2 -affected members of the same family, whereas in families with PSEN1 mutations, the age of onset is generally quite similar among affected family members, and it is even similar among people from different families with the same mutation5,51,91,116. Missense mutations in the PSEN2 gene may be of lower penetrance than in the PSEN1 gene and therefore may be subject to the modifying action of other genes or environmental influences51,117.
Gene location and structure The PSEN2 gene is located on chromosome 1 (1q42.13) and was identified by sequence homology and then cloned115,116. PSEN2 has 12 exons and is organized into 10 translated exons that encode a 448-amino-acid peptide. The PSEN2 protein is predicted to consist of nine transmembrane domains
Figure 43.3 AD3: PSEN1 structure and mutations. Thus far, there have been at least 123 mutations in the PSEN1 gene described, of which a few are shown. For a more complete list of PSEN1 mutations see www.molgen.ua.ac.be/ADMutations. TM: Transmembrane domains. Scale is approximate
Source:
Related posts:
 The Care Home Experience Alisoun Milne
The Care Home Experience Alisoun Milne
 The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
The Informant Questionnaire on Cognitive Decline in the Elderly (IQCODE) Nicolas Cherbuin andAnthony Francis Jorm
 The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
The Lundby Study Mats Bogren, Cecilia Mattisson and Per Nettelbladt
Environmental Factors, Life Events and Coping Abilities Toni C. Antonucci and James S. Jackson
The Multidisciplinary Team and Day Care Provision Martin Orrell and Kunle Ashaye
 Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Emerging Applications of Gene and Somatic Cell Therapy in Geriatric Neuropsychiatry Eric Wexler
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


