Global Vascular and Metabolic Injuries of the Brain
Juan C. Troncoso
Brain tissue has a high metabolic rate and therefore is susceptible to injury in conditions of ischemia, hypoxia, or metabolic derangements that impair the generation of energy (adenosine triphosphate) by neurons and other brain cells. To supply this high-energy requirement, the range of normal cerebral blood flow (CBF) is between 50 and 75 mL/100 g/min. Lower CBF results in depolarization of membranes and functional impairments that are reversible. This reversibility decreases as the CBF approaches 10 mL/100g/min, the threshold for neuronal cell death.1
Hypoxia and ischemia are different pathologic processes that often occur together. Whereas in hypoxia, only the supply of oxygen to the brain is impaired, ischemia also causes failure to remove toxic metabolic products (i.e., carbon dioxide and hydrogen). Thus, ischemia has more severe consequences than hypoxia. In fact, hypoxia alone causes synaptic and electrical failure, but lacks the glutamate release and neuronal necrosis of ischemic injury. This difference is important both to the clinician and the pathologist. Although conditions of pure brain hypoxia are rare, they may occur sometimes in acute respiratory disorders (i.e., asthma, epiglottitis, anaphylaxis). The patient, usually young, develops respiratory arrest without cardiac arrest and becomes comatose. The coma lasts for 1 or 2 weeks and, notably, it may be followed by complete neurologic recovery.2, 3, 4, 5 In contrast, total brain ischemia for as short as 2 minutes can cause neuronal necrosis in experimental animals.6 Although hypoxia per se does not cause neuronal necrosis, it does have the potential to enhance the ischemic injury.
In this chapter, we also use the term hypoxic-ischemic injury or encephalopathy, because although it is possible to separate hypoxia and ischemia in experimental situations, these two abnormalities usually coexist in clinical settings (with the exceptions noted above). Although the initial mechanism could be hypoxia alone (i.e., respiratory depression because of narcotic overdose), the thresholds for tissue injury of the brain and heart are very close, thus, when hypoxia reaches a level damaging to the brain, the heart will also stop beating or go into a ventricular arrhythmia leading to brain and, at times, also spinal cord ischemia.
In broad practical terms, the neuropathologist is confronted with four types of global injury of brain tissues: (1) complete global ischemic injury, as seen in cardiorespiratory arrest or ventricular fibrillation; (2) incomplete global ischemic injury, as seen in states of severe arterial hypotension; (3) global brain ischemia with preserved cardiorespiratory function, for example, neck compression; and (4) metabolic derangement, such as in severe hypoglycemia or toxicity with a mitochondrial poison such as cyanide. In recent years, significant progress has been made in the understanding of the mechanisms that lead to global cerebral ischemia; however, these mechanisms are complex and not thoroughly understood (for a review, see Harukuni and Bhardwaj1). One outstanding example of poorly understood pathogenesis is the occasional delayed leukoencephalopathy after hypoxic-ischemic injury.7 In cases of successful resuscitation, the re-establishment of CBF can lead to reperfusion injury because of increased reactive oxygen species, inflammatory changes, edema, and hemorrhage.
COMPLETE GLOBAL BRAIN ISCHEMIA RESULTING FROM CARDIORESPIRATORY ARREST OR VENTRICULAR FIBRILLATION
Complete global brain ischemia, also known as transient global ischemia or stagnant hypoxia, is a common scenario in forensic neuropathology.5 In our experience, the most common causes of cardiorespiratory arrest are heart diseases or an overdose with narcotics leading to respiratory depression and heart arrest. Unless the perfusion and oxygenation of the brain is re-established within a few minutes, neurons and other brain cells will be injured and eventually die. Clinical experience with extracorporeal circulation situations has shown that ventricular fibrillation or asystole for 5 to 10 minutes results in clinically manifest global cerebral ischemia.
The ischemic injury of the brain, depending on its severity, has consequences ranging from selective neuronal necrosis (only nerve cells damaged) to infarction (neurons, glia, and blood vessels involved). Furthermore, the threshold for ischemic injury varies for different brain regions, a phenomenon known as selective vulnerability. The most vulnerable regions are neurons in layer 3 of the cerebral cortex, globus pallidum, CA1 of the hippocampus, and Purkinje cells of the cerebellum. In our experience, great individual variability exists regarding which group of neurons is more vulnerable to ischemia. In general terms, the brainstem is more resilient to ischemia than the cerebrum and cerebellum. When ischemia is prolonged, however, brainstem and spinal cord suffer neuronal necrosis as well. Other important factors that modulate neuronal vulnerability to ischemia and hypoxemia are age and temperature. The brains of younger individuals are more resistant to ischemia than those of older subjects; lower temperature is also a protective factor. As a result, children who have been submerged in cold water for relatively long periods of time can be resuscitated with minimal or no neurologic deficit.8, 9 Hypothermia is used to protect the brain from ischemic injury in cardiovascular surgical procedures that require total circulatory arrest
(i.e., correction of tetralogy of Fallot and aortic arch surgery).10 Furthermore, clinical trials have shown that induced hypothermia reduces neurologic impairments and mortality after cardiac arrest.11, 12
(i.e., correction of tetralogy of Fallot and aortic arch surgery).10 Furthermore, clinical trials have shown that induced hypothermia reduces neurologic impairments and mortality after cardiac arrest.11, 12
The severity of the pathologic changes after hypoxemic-ischemic encephalopathy depends on the duration and intensity of the hypoxemic-ischemic injury. The range of lesions extends from necrosis of a few neurons in the most susceptible regions (Fig. 11.1) to pseudolaminar necrosis of the cerebral cortex (Figs. 11.2A and B) to global infarct of the brain (Fig. 11.3). The detection of these changes, however, depends on the lapse of time between injury and death. The brains of individuals who die immediately or within a few hours after suffering hypoxic-ischemic encephalopathy may appear normal on gross and light microscopic examination. After 24 hours between injury and death, the gross examination is characterized by edema and dusky discoloration of the cortical ribbon and cerebellar folia (Fig. 11.4). In addition, the normal sharp demarcation of the cerebral cortex and underlying white matter becomes progressively blurred. With the passing of time, all of these features become more pronounced, and the marked edema may cause uncal or tonsillar herniation. Lesions of the mammillary bodies, brainstem, and spinal cord are not uncommon after cardiac arrest. These lesions are usually symmetric (Fig. 11.5).
The microscopic hallmark of hypoxemic-ischemic injury is the necrotic neuron, characterized by a shrunken nucleus and an eosinophilic perikaryon with effacement of organelles, in particular, the rough endoplasmic reticulum (Fig. 11.1). The accepted mechanism for neuronal necrosis is excitotoxicity triggered by excitatory amino acids, such as glutamate. Apoptotic neurons are exceptional in the adult brain after ischemic injury but are occasionally present in the brain of fetuses and newborns. In experimental settings, prominent neuronal apoptosis of the granule cells of the hippocampus has been observed in dogs undergoing hypothermia and total circulatory arrest within 6 to 12 hours after the procedure, followed by neuronal necrosis evident 1 or 2 days later. Depending on the severity of the injury and the time transpired, the number of necrotic neurons in human cases varies from a few to many. In contrast with infarcts, even in severe hypoxemic-ischemic injuries, a few neurons are always spared. The intervening neuropil appears edematous initially and by the third or fourth day the reaction of astrocytes becomes evident. A macrophagic response followed by prominent proliferation of reactive astrocytes will develop if there is substantial neuronal death and tissue injury, and its chronology is similar to that of infarcts13 (Fig. 11.6). For all these reactive changes to occur, perfusion of the brain must be re-established. For this reason, foci of hemorrhage are sometimes encountered amidst the dead neurons, a phenomenon attributed to reperfusion of damaged capillary beds14 (Fig. 11.7).
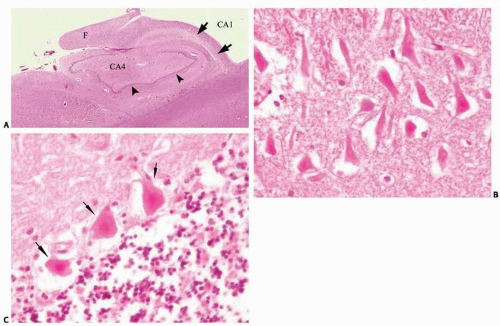 FIG. 11.1. Necrotic neurons are the morphologic hallmark of ischemic injury. On hematoxylin-eosin stains, ischemic neurons have an intense pink or red discoloration, the perikaryon appears homogenized as a result of the loss of the endoplasmic reticulum, and the nucleus becomes markedly pyknotic. This figure illustrates lesions in two brain regions selectively vulnerable to hypoxia and ischemia. A. Hippocampal formation with pallor of the CA1 region reflecting the loss of neurons (arrows) several weeks after cardiac arrest. The arrowheads mark the fascia dentata and the “F” the fimbria. B. Necrotic pyramidal neurons of CA1 region of the hippocampus 3 days after cardiac arrest. C. Acute ischemia of the cerebellum: arrows point at three necrotic Purkinje neurons.
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|





