Fig. 56.1
Neurosarcoidosis. (a) Sagittal T1-weighted precontrast MR image. (b) Sagittal T1-weighted gadolinium-enhanced image. (c) Axial T1-weighted gadolinium-enhanced image. On (a) and (b), the pituitary stalk and gland are diffusely enlarged, with relatively homogeneous enhancement. On (c), the cisternal segments of the cranial nerve V demonstrate enhancement
A considerable contrast enhancement pattern of granulomatous component is typical, which may respond to treatment [19].
Thickening of the infundibulum and attenuation of the posterior pituitary bright spot are frequently associated [20, 21].
Lesions may be solid or cystic, depending on the contents and degree of necrosis [17].
56.1.3 Histopathology
The histopathology of sarcoidosis involving the hypothalamic–pituitary region is similar to that in other systemic locations, characterized by collections of well-formed, noncaseating granulomas admixed with a moderate amount of lymphoplasmacytic inflammatory infiltrates [23]. Aggregates of activated epithelioid histiocytes with foreign body–type or Langhans multinucleated giant cells are characteristic of the granulomas. The inflammatory infiltrate shows a predominance of CD4+ lymphocytes. Tissue destruction may be extensive, and chronic lesions may present extensive stromal fibrosis and even calcification (Fig. 56.2).
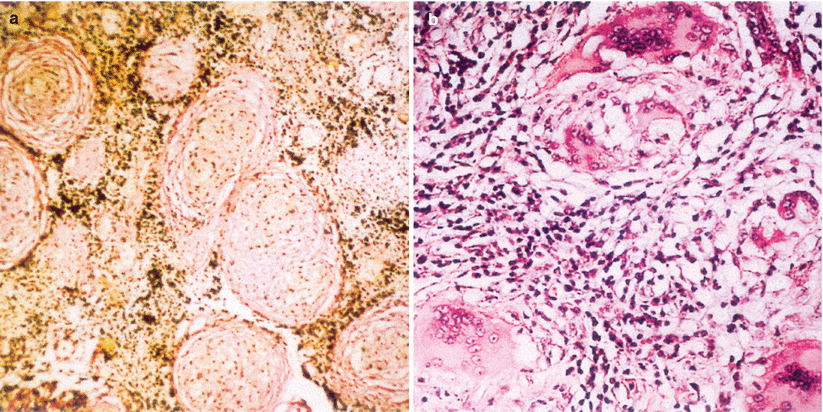
Fig. 56.2
Neurosarcoidosis. (a) Characteristic noncaseating sarcoid granulomas embedded within a fibrous stroma (Van Gieson, original magnification ×80). (b) Sarcoid granulomas consisting of follicles with central, multinucleated giant cells (some with polymorph, conchoid inclusion bodies) surrounded by epithelioid macrophages and fibroblasts (H&E, original magnification ×200) (Adapted from Nowak and Widenka [23], with permission)
The diagnosis of sarcoidosis may be difficult and requires the exclusion of other diseases (mainly infectious), by clinical and laboratory means. Histochemical stains for fungi and mycobacteria, microbiological cultures of the tissues, and/or PCR-based tests are mandatory. Likewise, microbiological studies of CSF and serum should be performed to exclude more systemic infectious causes.
Other causes of systemic granulomatous diseases (such as Churg–Strauss syndrome, lymphomatoid granulomatosis, and Wegener’s granulomatosis) should also be ruled out. Detailed clinical and laboratory correlation is significant for the differential diagnosis.
Involvement of the sella and suprasellar structures in Wegener’s granulomatosis is uncommon. Radiologically, the gland is diffusely enlarged and exhibits a more homogeneous pattern of enhancement [21].
Tuberculosis of the sellar and parasellar region is extremely rare. It has been reported to present in many ways, such as a thickened stalk, a hypophysitis with contiguous involvement of the stalk, a mass similar to a pituitary adenoma, and even a pituitary abscess.
Neurosarcoidosis can be differentiated from tuberculous granulomas or abscesses via caseating necrosis and positive acid-fast bacillus (AFB) cultures.
56.1.4 Clinical and Surgical Management
Surgical intervention, often via a transsphenoidal approach, can be utilized for biopsy or debulking, especially for drainage of cystic masses.
For any granulomatous lesions, tuberculosis should be ruled out with PPD and chest x-ray.
Upon diagnosis of a noncaseating granuloma without previously known systemic disease, a thorough workup for systemic conditions should be performed. For neurosarcoidosis, this workup includes a serum angiotensin-converting enzyme (ACE) level, erythrocyte sedimentation rate (ESR), calcium levels, gallium-67 lung scan, and bronchoalveolar lavage [24].
CSF studies also should be performed, including cytology, cell counts, and ACE levels [16].
Medical therapy typically includes corticosteroids and perhaps cyclosporine A or chloroquine [25, 26].
Up to 87 % of neurosarcoidosis lesions will improve with steroid treatment, especially in regard to demonstration of contrast enhancement [27].
Central diabetes insipidus and hypopituitarism typically fail to improve after treatment [16].
56.2 Wegener’s Granulomatosis of the Sellar Region
56.2.1 Epidemiology and Clinical Presentation
Wegener’s granulomatosis (WG), a type of autoimmune vasculitis of small- and medium-sized blood vessels, rarely has intracranial manifestations. It typically manifests in the lungs, skin, and kidneys, but in rare cases, WG has been described to involve the pituitary gland and parasellar region.
Pituitary WG is almost always diagnosed in the presence of other systemic findings [28].
The mean age of patients with pituitary WG is 38 years [28].
Diabetes insipidus and hypopituitarism are common features of WG involving the pituitary gland [4, 28–30].
Involvement of the cavernous sinus presenting with cranial nerve paresis has also been described [31].
Systemic manifestations of WG may also be evident, including fevers, rash, and pulmonary features [29].
56.2.2 Imaging Features
On MRI, WG of the pituitary gland typically appears as an enhancing mass that is hypointense on T1-weighted imaging and hyperintense on T2 imaging (Fig. 56.3) [28, 29, 32].
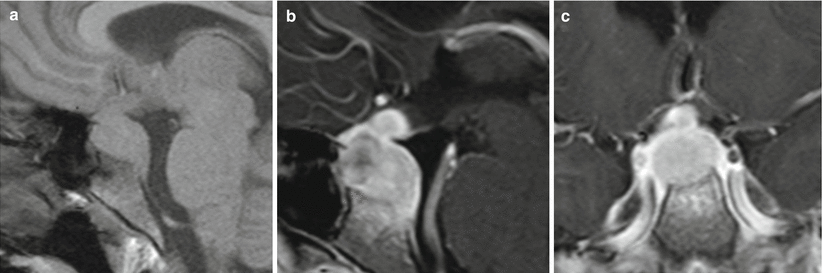
Fig. 56.3
Wegener’s granulomatosis. (a) Sagittal T1-weighted precontrast MR image. (b) Sagittal T1-weighted gadolinium-enhanced image. (c) Coronal T1-weighted gadolinium-enhanced image. The pituitary stalk and gland are diffusely enlarged, with homogeneous enhancement. On (b), erosion of the sellar floor and enhancement of the upper clivus are visible
MRI also may show extension from the paranasal sinuses into the sella and cavernous sinus, often with meningeal thickening [31].
Infundibular thickening and cystic changes also may be seen on MRI [28].
Loss of the normal posterior pituitary bright spot may be seen, especially in patients with WG and diabetes insipidus (Fig. 56.4) [21, 32].
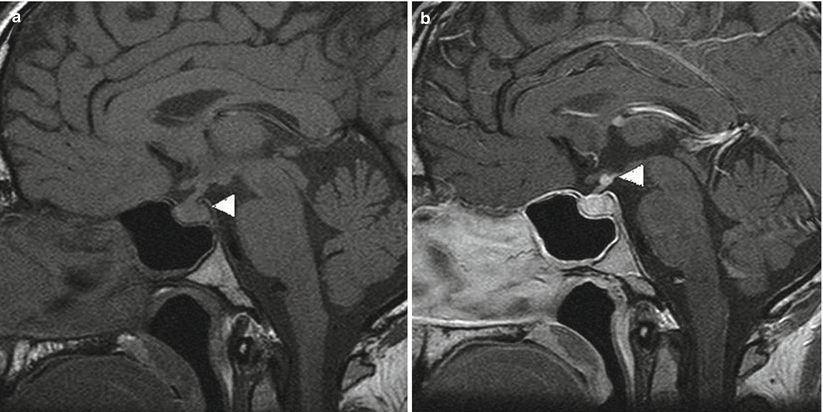
Fig. 56.4
Wegener’s granulomatosis. (a) An unenhanced T1-weighted sagittal MR image through the pituitary gland shows the absence of the “bright spot” of the posterior pituitary (white arrowhead), typically seen with central diabetes insipidus. (b) A post-gadolinium T1-weighted sagittal image shows an enhancing nodule at the superior aspect of the infundibulum (white arrowhead) (Adapted from Yong et al. [28], with permission)
The disease may be characterized by exacerbations and periods of quiescence during which MRI findings may appear essentially normal [21].
56.2.3 Histopathology
WG is characterized by inflammation of the small blood vessels (polyangiitis).
Granulomatous necrotizing inflammation of the pituitary gland can be seen [33].
The disease is characterized histologically by an inflammatory infiltrate comprising neutrophils, lymphocytes, plasma cells, histiocytes, and eosinophils.
56.2.4 Clinical and Surgical Management
The diagnosis of WG can be made by confirming c-antineutrophil cytoplasmic antibodies (c-ANCA) and positive skin lesions showing leukocytoclastic vasculitis [29].
ANCA is positive in 80–95 % of cases [28].
Proteinase 3 (PR3) positivity is seen in 70–80 % of ANCA-positive patients [28].
The role of surgery is minimal in this disease; it may be limited to biopsy and/or sellar decompression.
Systemic treatment often includes high-dose corticosteroids and immunosuppressive medications such as cyclophosphamide. Most patients respond favorably to cyclophosphamide, although one review reported relapse in 27 % of patients [28].
The monoclonal antibody rituximab has recently been used successfully to treat refractory pituitary WG [34].
Hypopituitarism and diabetes insipidus rarely improve after remission of pituitary WG, making vigilant endocrinological follow-up imperative [28].
56.3 Langerhans Cell Histiocytosis
56.3.1 Epidemiology and Clinical Presentation
Langerhans cell histiocytosis (LCH), previously called histiocytosis X, is a clonal neoplastic proliferation of Langerhans-type histiocytes that typically develops in childhood.
The estimated incidence of LCH in children is 1–5 per million per year [35].
In children, the median age at diagnosis is 30 months.
LCH may present in young adult patients (mean age 34 years), typically with diabetes insipidus and hypopituitarism [36].
LCH is more common in males overall, but isolated hypothalamic–pituitary involvement may be more common in females [37].
The wide spectrum of clinical presentation includes many syndromes [38]:
Eosinophilic granuloma: unifocal or multifocal lytic bony disease
Hand–Schuller–Christian disease: diabetes insipidus, exophthalmos, and bony lesions
Letterer–Siwe disease: fulminant disease occurring in infants
Most children present with monostotic (61 %) or polyostotic lesions (27 %).
Isolated skull lesions are the most common presentation.
A unifocal hypothalamic–pituitary lesion (Gagel granuloma) is the presentation in 5 % [38].
LCH granulomas comprise approximately 0.05 % of cases in major transsphenoidal series [39].
The most common (and often the first) manifestation is central diabetes insipidus, occurring in 11–25 % of patients with LCH [37, 40–42].
Anterior pituitary dysfunction occurs in up to 24 % of patients overall, almost always in association with diabetes insipidus [37, 43].
Growth hormone (GH) deficiency develops in more than half of patients with diabetes insipidus [44].
56.3.2 Imaging Features
LCH patients with sellar region findings on MRI often present with:
Thickening of the pituitary stalk (50–86 %) (Fig. 56.5) [45, 46]
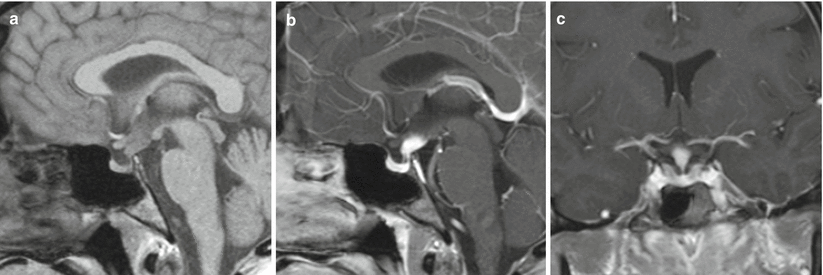
Fig. 56.5
Langerhans cell histiocytosis. (a) Sagittal T1-weighted precontrast MR image. (b) Sagittal T1-weighted gadolinium-enhanced image. (c) Coronal T1-weighted gadolinium-enhanced image. The pituitary stalk demonstrates nodular enhancement superiorly. The pituitary gland is normal in size
Suprasellar granuloma: typically isointense on T1-weighted images and hyperintense on T2-weighted images (10 %) [47]
Intracranial involvement can be multifocal, frequently involving the cerebellum with enhancing or nonenhancing signal abnormalities.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree








