Title
Authors
Scale description
Functional Capacity Rating Scale
Shoulsan and Fahn (1979)
Measures functional capacity across 5 domains on a scale of 1–5
Unified Huntington’s Disease Rating Scale (UHDRS)
Kieburtz (& Huntington’s disease study group; 1996)
A multi-domain measure of disease progression across 6 domains of function. Includes a Functional Assessment and Total Functional Capacity Scale
Core Assessment Program for Intracerebral Transplantation in Huntington’s disease (CAPIT-HD)
Quinn et al. (1996)
A multi-domain assessment protocol originally developed for the transplantation program
The course of Huntington’s disease can nevertheless be variable. The average age of motor onset is around 42 years but HD can begin in childhood (Juvenile HD or Westphal variant) or even in the elderly. The extremes of onset age are determined in large part by the inherited CAG repeat length. Juvenile onset (onset before 20 years of age) occurs in about 5–15 % of HD cases worldwide, and presents with a bradykinetic form of the disease, which appear Parkinsonian and they may have seizures. Generally, the first signs are related to a drop in school performance. In rare pedigrees, parents of affected individuals may not have clinical evidence of disease despite living to an advanced age.
CAG repeat lengths lower than 26 are considered stable and result in no clinical symptoms or known increased risk to future generations. Repeats of 27–35 CAG repeats are considered intermediate, and symptoms are not expected to manifest in these individuals but CAG repeats of this length are ‘unstable’ and can lead to greater expansions with morbidity in future generations. Repeat lengths of 36–39 CAG repeats have reduced penetrance, with most individuals becoming symptomatic, but often with a milder expression in midlife. In adults, the length of the CAG repeat expansion accounts for up to 70 % of the variability relative to age of onset. Childhood onset of HD is usually caused by an especially large CAG repeat (i.e., >50). By contrast, length of the CAG repeat appears to contribute less to the rate of disease progression.
The individual with early signs of HD has grown up in a family in which one of the parents likely became affected in adulthood and may have passed through the course of the disease and died. The newly affected individual’s family life may have been severely disrupted during their childhood by the parent’s change in behaviour. The psychological stress in such individuals is easy to underestimate. They may be living with a sense of dread about the insidious onset; with even minor neurologic complaints being interpreted as disease expression for instance benign myokymia is often misinterpreted as the onset of chorea. A simple mechanical fall or stumble can develop an elaborate significance it does not warrant. In the disease’s prodromal phase, depression can develop years before the onset of motor symptoms, and the highest prevalence has been reported within 1 year of clinical diagnosis.
9.3 Prevalence
In the general population of the western hemisphere, the prevalence rate of HD is estimated to be 4–10 per 100,000. There is large variance in prevalence ratings, though a 20-year retrospective analysis of records in the UK recently revealed the prevalence rate has risen from 5.4 per 100,000 in 1990 to 12.3 per 100,000 in 2010. A higher prevalence has been reported in South Wales and Venezuela as compared to a lower rate found in Finland, Japan and African-American populations. This variability is due, predominantly, to the relative mobility of carriers of the gene and the existence of isolated “pockets” of families living in close proximity.
9.4 Pathology
Huntington’s disease causes neuronal loss starting in the striatum but eventually leading to progressive whole brain atrophy. Early in the disease there may be little or no signs of the disease in the gross brain. However, inevitably neuronal loss develops with atrophy of the cortical surface in most and of the striatum in nearly all cases where the neuronal loss is accompanied by astrocytosis. There is a particularly severe degeneration of the caudate nucleus that begins in the tail of the caudate and then advances in predictable way to the dorsal medial aspect then to the ventrolateral side. Eventually the caudate atrophies to a thin tissue paper-like gliotic structure that is devoid of usually predominating medium spiny neurons. This gives a boxcar appearance of enlarged lateral ventricles on CT scan. The putamen is also affected in a predicable caudal to rostral vector whereas the nucleus accumbens is generally spared. Vonsattel described the standard approach to brain assessment in HD and used 1–4 grading system to standardize the pathological description from mild to severe involvement. In grades 3–4 there is much more widespread brain degeneration with cortical loss, white matter loss and extensive gliosis (see Fig. 9.1). Like many neurodegenerative disorders, Huntington’s disease is associated with abnormal accumulation and misfolding of the proteins. A role of non neuronal cells is also gaining support, in Huntington’s disease as well as in ALS genetic animals with the mutation in microglia or glial cells but not neurons is also associated with pathologic changes.
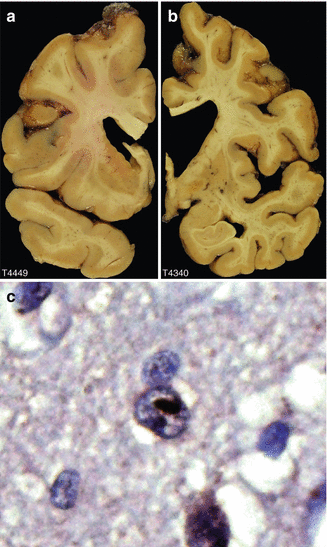
Fig. 9.1
Neuropathology of Huntington’s disease. (a) The caudate nucleus bulges into the ventricle in normal individuals but is flattened in this 55 year old person dying with Huntington’s disease as seen in this coronal section through one brain hemisphere. Characteristically the atrophy is more severe in the dorsal aspects of the caudate. (b) The caudate is barely visible in this 75 y.o. with severe degeneration of the caudate and globus pallidus. (c) The centre neuron contains a classic intranuclear inclusion composed of aggregates of the mutant huntingtin protein (Figures courtesy of Dr. Jean Paul Vonsattel of Columbia University)
9.5 Symptomatology
Huntington’s disease is associated with a triad of difficulties including the movement disorder as well as cognitive and neuropsychiatric conditions. Each of these is associated with a complex set of psycho-social problems. Patients with Huntington’s disease face a range of difficulties from diagnosis to death and these difficulties are not confined to the patient themselves but also to the family. As an autosomal dominant disorder the disease has a particular resonance with families of sufferers.
9.5.1 Movement Disorder
By the time that chorea manifests, Huntington’s disease generally includes some alteration in voluntary motor control. The ability to make rapid, repetitive, sequential movements is often abnormal.
Tests such as alternately tapping the thumb against the tips of the fingers, repetitively tapping the tip of the tongue against the top lip, alternately tapping the top, then the palm of one hand against the palm of the other hand all show slowing and irregularities in timing. There is generally great difficulty keeping the tongue protruded over a short, i.e., 10 s period.
Eye movement abnormalities are common. These include inability to make smooth pursuits due to intrusive saccades and delays in initiation of saccades. There is also dramatic slowing of saccadic velocity in some patients.
The gait of the person with early Huntington’s disease demonstrates increased variability in step length and distance from the intended path. Inability to maintain position after gently pulling the patient backwards, and trouble performing tandem walking is common. Though the cerebellum is generally spared, the finger to nose test and heel to shin test generally shows dysmetria.
Chorea, from the Greek for dance, often starts as a quick flick at multiple joints in the fingers or fingers and wrist while walking. It commonly worsens to twisting turns of the limbs, involuntary neck and facial movements, with blinks, and writhing tongue movements. The progressive involvement of lingual and bulbar control leads to dysarthria and dysphagia. Food with a soft moist consistency such as pudding is easiest to swallow. Maintaining adequate nutrition can be challenging and motor symptoms tend to worsen as patients lose weight. Most become almost mute in the later stages of the illness and are completely unable to swallow without aspirating. Chorea, which can be of large amplitude and forceful enough to cause self injury, tends to slow over years and the involuntary movements evolve to dystonia in the later stages of the illness. Walking becomes more and more associated with falls and eventually the person is wheelchair or bed bound. Tone increases with disease progression. There is commonly a dramatic, reflexive increase in tone when the limb is activated. Reflexes are hyperactive. The Babinski reflex often becomes positive.
In contrast to the major motor findings, sensory abnormalities are minimal or absent. Some believe that patients with advanced HD have decreased pain sensation.
9.6 Cognition
Many of the early studies reported general intellectual and cognitive decline in HD, which is worse than in other neurodegenerative conditions. However, intellectual ability generally remains stable over time, with the most pronounced deficits seen in executive domains in parallel with pathological changes. A recent meta-analysis of the literature, which incorporated the results from 760 patients, showed maximum differences between controls and HD patients, using the effect size statistic, were found on tests of construction, and memory.
In a more recent population based cross-sectional study of motor manifest HD gene-expansion carriers, 51.8 % presented with the full symptom triad i.e., cognitive, neuropsychiatric, and motor involvement, 25 % were defined as cognitively impaired in addition to motor symptoms, and 14.3 % had neuropsychiatric symptoms along with motor symptoms. Only 8.9 % had isolated motor symptoms. Among the HD participants without motor symptoms, 39.2 % had neuropsychiatric symptoms, were cognitively impaired, or had a combination of both. Cognitive decline precedes motor signs in many gene-positive HD patients, and may be an important target in clinical trials and early intervention. Cognitive test scores may also improve the ability to predict disease onset among gene mutation carriers and help families to better plan for potential personal and economic strain.
In a recent observational report over a 36-month period of 366 participants with HD, the symbol digit modality test proved to be especially sensitive to cognitive change. Among psychiatric indicators, apathy ratings specifically showed significant increases compared to controls. Several baseline imaging, quantitative motor, and cognitive measures had prognostic value, independent of age and CAG repeat length, for predicting subsequent clinical diagnosis in pre-HD.
Cognitive reserve has been shown to have positive prognostic value in HD, comprised of composite scores derived from estimated premorbid intellectual level, occupational status, and years of education, relative to disease progression in HD. Higher cognitive reserve was significantly associated with a slower rate of change and slower rate of volumetric loss in two brain structures (caudate, putamen) for those estimated to be closest to motor disease onset. These findings demonstrate a relationship between cognitive reserve and both a measure of executive functioning and integrity of certain brain structures in pre-motor HD individuals.
Diagnostic algorithms have gained more attention in recent years in HD, where CAG-expansions are computed to obtain an estimated “years to clinical diagnosis”. Stratifying groups as: near, estimated to be 9 years from diagnosis; mid, between 9 and 15 years; and far, >15 years, researchers conducted neurocognitive assessments on each cohort. Nineteen cognitive tasks were used to assess attention, working memory, psychomotor functions, episodic memory, language, emotional recognition, sensory and perceptual functions, and executive process. The near group showed significantly poorer performance on nearly all of the cognitive tests, and the mid group on about half of the cognitive tests. Overall, the cognitive battery accounted for 34 % of the variance. This further highlights how neurocognitive tests are robust clinical indicators of the disease process prior to reaching criteria for motor diagnosis of HD. Six main cognitive factors have to be considered when monitoring markers of disease progress, and cognitive indices should be used when monitoring cognitive function in pre-motor HD, rather than results from individual measurements or screening tools. These factors include: (1) speed/inhibition, (2) verbal working memory, (3) motor planning/speed, (4) attention-information integration, (5) sensory-perceptual processing, and (6) verbal learning/memory. Overall, motor planning/speed and sensory-perceptual processing appear to be the most important markers of disease prognosis.
Thus many studies of cognitive functioning in HD report generalised decline in cognitive functioning over the course of the condition. However, the specific nature of this decline varies between studies. It is likely that this reflects an inherent variation in the presentation of the disease within and between families as well as poor study design and differing measures. In particular, because of the relative rarity of the disease, many studies are insufficiently powered with respect to patient numbers.
9.6.1 Language Ability
Traditionally, HD patients do not present with clear cortical aphasia. However, as more sophisticated testing has emerged it has become clear that many patients presented with a range of language based functions, some of which are masked by the severity of dysarthria. Comprehension is generally thought to be intact. However, HD patients have been shown to be impaired in the comprehension of affective and propositional (command or question) prosody.
9.6.2 Executive Functions
Recently, there has been increased interest in the executive deficits experienced by patients with HD. This not only results from the earlier application of more sophisticated diagnostic testing that enables patients to complete tests sooner in the course of the disease, but also reflects the improved resolution of current neuroimaging techniques. Such techniques have facilitated a greater description of the nature of the lesions in HD and identified degeneration in the frontal lobes via fronto-striatal connections. That is, the nature of the reciprocal connections between the basal ganglia and the oculomotor region, dorsolateral prefrontal cortex and lateral orbitofrontal areas resulting in significant degeneration in fronto-striatal mediated cognitive and behavioural functions.
HD patients have been shown to be impaired on tests of planning, self-order working memory and tests of response set, all indicators of executive dysfunction. Although HD patients have been shown to perform poorly on measures of categorical fluency, more deficits are prevalent on phonological fluency (letter) rather than semantic fluency (category). Consistent with many studies, patients with a more rapidly progressive prognosis (determined using mutation size and current age) perform more slowly and with less accuracy on tests of executive function. Performance accuracy tends to be negatively related to striatal volume while both accuracy and working memory are negatively related to frontal white matter volume. Interestingly, pathological disturbances in cortico-striatal circuits in HD present similarly to other pathologies such as excessive gambling. Although similar disinhibition related symptoms are present i.e., changed sensitivity to punishments and rewards, impulsivity, and inability to consider long-term advantages over short-term rewards, both HD patients and pathological gamblers also show similar performance deficits on risky decision-making tasks and measures of executive function.
In summary, patients with HD show deficits on a range of tests of executive function. These take the form of planning, executing and inhibiting behaviour. In the context of the neuropathological data it is not surprising that HD patients should present with such difficulties. Loss of frontal white matter and neuronal cell loss have been reported in many studies and metabolic deficits in the frontal lobes have been associated with the degree of cell loss in the basal ganglia.
9.6.3 Attentional and Perceptual Functions in HD
In many studies, HD patients were found to be impaired on tests of alertness, divided attention and response flexibility. While clear disorders of perception are rare, there has been increasing interest and controversy surrounding the issue of patient’s ability to perceive emotional cues. HD patients were impaired at interpreting facial and vocal expressions of emotion, and similarly, those relating to fear and disgust were disproportionately impaired. HD patients have also been shown to be impaired at comprehending emotional prosody in speech, matching facial affect, facial recognition and discriminating faces.
9.6.4 Memory
There has been a great deal of debate concerning the nature of the memory impairment in HD and the pattern of impaired and preserved skills in this patient group. It has been clear for many years that patients present with a form of memory impairment that, while severe, is distinct from other dementias such as Alzheimer’s disease.
Global memory deficits are common in HD and this is unsurprising given the links between the striatum and limbic system, the reported deficits in temporal lobe function and the deficits in frontal function in this population. Unlike Alzheimer’s patients where episodic memory is better for older memories, HD patients show no advantage for older memories over more recent ones. This is known as a flat temporal gradient. Procedural learning is also impaired. In particular, procedural motor tasks are more impaired than lexical tasks. It has been suggested that recognition memory is disproportionately preserved until later stages and therefore retrieval or encoding deficit are favoured by many authors. That is, the memory impairment in HD, which begins early in the course of the disease, is related to the more extensive and global deterioration in fronto-striatal functions.
9.6.5 Social Cognitive Processes
Multiple reports suggest that HD patients (in both manifest and preclinical stages) have multimodal deficits in emotional processing that extend to decoding facial expressions, prosody, body language and rating of emotional scenes, with some but not all studies suggesting that negative emotions such as disgust and anger are disproportionately affected.
The term “Social Cognition” encompasses several sub-domains including emotion recognition from facial expressions, prosody or body posture, as well as, the ability to infer other people’s mental states in terms of beliefs, desires, feelings, intentions, or knowledge states.
Patients with HD have been reported to make significantly more errors on measures of social cognition than controls, exhibiting difficulties in judging the social appropriateness of character’s behaviour in stories, and problems inferring complex mental states through commonly used measures (the use of photographs of people’s eyes). HD patients have further evidenced lower everyday perspective taking scores relative to a matched sample. Social cognitive deficits have been shown to appear independent of executive dysfunction, however, executive deficits are linked to poor understanding of socially inappropriate remarks and errors in mental state attribution.
HD patients have notable deficits in their ability to identify fear, disgust, and anger. These results are the most consistently reported, on non-demanding tasks, and across modalities (i.e., visual and auditory). Declines in emotional recognition exist regardless of whether the emotions are being communicated by others’ facial expressions or vocal intonation, although patients’ emotional recognition deficits are not necessarily evident in, or associated with, increased functional problems with social interactions. This finding points to a differential impairment in emotion recognition and emotional experience in HD.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


