(1)
Asheville, NC, USA
The year 2001 witnessed the publication of the human genome sequence by two consortia, one headed by Eric Lander (in Nature) and the other led by Crag Venter (in Science). The number of protein coding sequences, in the 20,000–30,000 range, and their chief characteristics, were reported. The coding sequences accounted for no more than about 1.5 % of the total genome. Both reports discussed this dramatic finding in detail and presented an overview of the entirety of the genome. In Lander’s report, repeat sequence s were noted as spanning 50 % of the genome. That such sequences are present was known for some time and made famous under the rubric “selfish DNA” or “selfish genes.” These sequences were divided into five classes by the Lander-led team—(1) transposon-derived repeats; (2) processed pseudogenes; (3) simple sequence repeats; (4) segmental duplications—blocks of DNA copied from one place to another, and (5) blocks of tandem-repeated sequences such as those encountered at centromeres and telomeres. The first group, transposon-derived repeats, constituted the bulk of the repeat content comprising 45 % of the genome. The third group, simple sequence repeats, consisted of repeated doublets such as (CA) n , triplets such as (CGG) n , repeated quartets, and so on. These repeats, also known microsatellites, accounted for 3 % of the genome.
That the vast bulk of repeat-laden DNA is not useless, inert, or entirely benign has become increasingly apparent during the past few years. One of the chief examples of this aspect is the role that unstable simple sequence repeats have in human disease. At least 30 neurodegenerative disorders are due to them. These diseases include Huntington’s disease, a variety of ataxias , monotonic dystrophy, the most common form of autosomal dominant muscular dystrophy, and fragile X syndrome , a leading form of inherited mental retardation. Up to now the main type of disease-causing mutations resulting in protein misfolding was the point mutation. Here, another kind of mutation is considered, one in which the number of simple sequence repeats increases from generation to generation from a normal number of repeats which may number up to 50, to disease-associated ever larger numbers ranging into the thousands. These increases correlate with an increasing severity of the illness, a phenomenon known as genetic anticipation .
Nine neurodegenerative disorders are known to be caused by unstable CAG repeat s situated in protein coding regions. These repeats encode the amino acid glutamine, hence the name polyQ diseases. These neurodegenerative disorders are listed in Table 10.1. Huntington’s disease heads this list. It is caused by unstable repeats in the huntingtin gene (HTT) situated on the short arm of chromosome 4 at location 4p16.3. This gene encodes the huntingtin (Htt) protein. Individuals possessing expanded numbers of these repeats develop Huntington’s disease, a fatal disorder that affects neurons in the brain resulting in motor dysfunction and progressing to cognitive decline, dementia, and death 15–20 years after onset of the illness. The primary targets are the medium spiny neurons (MSNs) situated in the striatum that deteriorate and eventually die off. These neurons receive cortical and thalamic input and release the inhibitory neurotransmitter γ-aminobutyric acid (GABA) to multiple regions involved in motor control. Like the illnesses discussed earlier, this disease spreads to other regions of the brain, in this case, to neurons in the hippocampus and cortex. Incidence of this disease among white populations is roughly 5–7 individuals per 100,000.
The first of the CAG repeat disorders to be discovered was that of the androgen receptor which gives rise to spinal and bulbar muscular atrophy (SBMA), also known as Kennedy disease. The disease was first described by William Kennedy and coworkers in 1968 but it was not until 1991 that La Spada and colleagues isolated the cause of the disease as the presence of expanded CAG repeats in the gene encoding the androgen receptor. The gene is located on the long arm of the X-chromosome, and the mutation is inherited in an autosomal recessive manner. Later in the same year, 1991, a pair of papers appeared reporting that Fragile X syndrome arises as a consequence of an unstable CGG repeat in the FMR1 gene, also located on chromosome X. In this case, the repeat expansion occurs in the 5ʹ UTR (untranslated region). That finding answered what had been called the Sherman paradox —the until-then unexplained increases in the disease susceptibility among successive generations observed in the mid-1980s by Stephanie Sherman.
Neurons in several areas of the brain are affected in the CAG repeat disorders. Prominent among these regions are spinal motor nuclei, brainstem nuclei, basal ganglia, and cerebellum. The spinocerebellar ataxias are the most common form of ataxia, a class of disorders that affect more than 150,000 people in the USA alone. Recall that the cerebellum coordinates muscle movement and regulates motor learning and control. Six of the nine entries in Table 10.1 are of spinocerebellar ataxias . Each of these illnesses affects a different portion of the cerebellum and each one is due to unstable expanded CAG repeats in a specific protein. The clinical features of each of the polyQ diseases are distinct, and depend crucially on repeat length and upon the biophysical properties and normal cellular functions of the affected protein. They do, however, share a number of notable features. One of these is the aforementioned increases from generation to generation in the severity of the illnesses due to the expanding number of unstable repeats. Another is the widespread appearance of neuronal intranuclear inclusions (NIIs).
Table 10.1
Polyglutamine (polyQ) repeat disorders
Neurological disease | Protein | Normal repeat length | Disease repeat length | Protein function |
|---|---|---|---|---|
Huntington’s disease (HD) | Huntingtin (Htt) | 6–34 | 36–121 | Transcription, transport |
Spinal and bulbar muscular atrophy (SBMA) | Androgen receptor | 9–36 | 38–62 | Nuclear receptor |
DentatoRubral and PallidoLuysian atrophy (DRPLA) | Atrophin-1 | 7–34 | 49–88 | Transcription |
Spinocerebellar ataxia 1 (SCA1) | Ataxin-1 | 6–39 | 40–82 | Transcription |
Spinocerebellar ataxia 2 (SCA2) | Ataxin-2 | 15–24 | 34–200 | RNA metabolism |
Spinocerebellar ataxia 3 (SCA3) | Ataxin-3 | 13–36 | 61–84 | Deubiquitination |
Spinocerebellar ataxia 6 (SCA6) | α1A-VDCC subunit, and α1ACT | 4–18 | 19–33 | P/Q type VDCC subunit |
Spinocerebellar ataxia 7 (SCA7) | Ataxin-7 | 4–35 | 36–460 | Transcription |
Spinocerebellar ataxia 17 (SCA17) | TATA-box binding protein (TBP) | 25–42 | 47–63 | Transcription |
Unlike the situation encountered in Parkinson’s disease where an impressive number of risk factor s and disease pathways have been uncovered, and where there are both familial and sporadic forms, HD arises from a single causative factor—mutations in the huntingtin protein encoded by the HTT gene. For that reason it might have been thought that HD and the other polyQ disorder s are straightforward, and easy to understand and treat. However, the huntingtin protein is large and remarkably complex. Like Alzheimer’s and Parkinson’s diseases, the underlying disease mechanisms and progression are neither simple nor easy to uncover. Huntingtin contains large stretches of unstructured sequences, is cleaved into different-sized fragments, is encountered with different repeat lengths, and is subject to a variety of posttranslational modifications.
Significantly, HD shares a number of biophysical and cellular features with many if not most of the other polyQ disorder s , and with AD and PD. Chief among them are transcriptional and axonal transport failures, loss of calcium homeostasis, dysregulated mitochondria, all leading to the loss of protein quality control , synaptic failure, and neuron death. Their exploration in this chapter begins with a look at the unusual, problem-causing secondary structures generated by the disease-causing expanded repeats. The biogenesis, membrane localization, and clearance of mutant htt follow with special attention to the central role exon 1 in causing Huntington’s disease. The exploration of Huntington’s disease concludes with an examination of huntingtin’s normal functions and disease-causing roles in intracellular vesicular trafficking , synaptic function, and mitochondrial biogenesis.
Those discussions are followed by an examination of the ataxias and the second major class of unstable repeat disorders, namely, those involving expanded repeats in non-protein-coding regions. This second group of disorders includes, besides fragile X syndrome , monotonic dystrophy 1 and 2, Friedrich’s ataxia, and several prominent forms of mental retardation. Study of these diseases will recapitulate one of the major themes of the polyQ class, the sequestration of proteins leading to their loss of function, and introduce a new mechanism—splicing errors—that makes a bridge to the RNA-centric disorders that will be encountered in the next and final chapter of the textbook.
10.1 Unusual Secondary Structures Underlie Trinucleotide Repeat Disorders
Instabilities in repeat lengths have their origin in the unusual secondary structure s they generate. Several different kinds of exceptional secondary structures are formed by the expanded repeats. Depending on the composition of the repeats and the process being undertaken these structures may involve ssDNA, dsDNA, or triplex DNA; others are established by DNA–RNA hybrid complexes and still others by RNA. Some will form hairpin-like protuberances, while others form loops, out-of-register, and slipped-strand realignments. These structural elements produce kinetic trap s, become “sticky” DNA, and cause numerous difficulties in replication, repair, and recombination. Once the number of repeats passes a certain threshold, they lead to cancers and neurodegenerative diseases. The different kinds of aberrant structures and the problems they cause are summarized in Fig. 10.1.
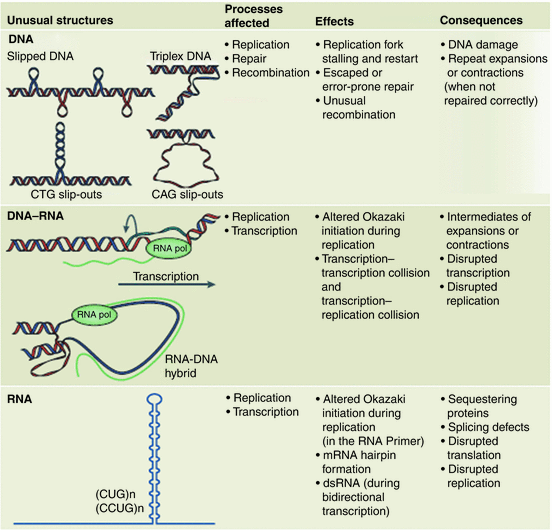
Fig. 10.1
Unusual problem-causing secondary structures generated by expanded repeats. Upper panel: slipped DNA, triplex DNA, and CTG and CAG slip-outs. Middle panel: DNA-RNA hybrid looped structures. Lower panel: RNA hairpin s . Columns 2, 3, and 4 list the most commonly affected processes, mechanistic effects, and ultimate consequences associated with these aberrant structures (from Castel Nat. Rev. Mol. Cell Biol. 11: 165 © 2010 Reprinted by permission from Macmillan Publishers Ltd)
One of the first theories on how these secondary structure elements might produce generation-to-generation increases in repeat length was the replication model. The central idea behind this model was that the unusual secondary structure s formed by the repeats act as impediments to replication and cause replication forks to slip and stall at those locations. Cells are equipped with a number of repair systems that fix damaged DNA and restart stalled replication forks. These factors, most notably the MSH2/MSH3 proteins that comprise the mismatch repair (MMR) recognition complex, and the base-excision repair (BER) 8-oxoguanine glycosylase (OGG1), are implicated in the repeat expansions. In addition, there is evidence for the involvement of recombination factors, cis acting elements such as the multifunctional transcription regulator CTCF, and epigenetics. The relative contributions of each of these factors to the disease etiology depend critically on the biophysical and biochemical properties of the repeats and cell-type under consideration. In carrying out the repair, additional repeat elements become inserted. In Fig. 10.1 a variety of consequences during replication, repair, recombination, and transcription have been listed.
10.2 Structure of the Huntingtin Protein
The 3144-amino-acid-residue huntingtin protein is organized into N- and C-terminal domains (Fig. 10.2). As noted earlier the protein is both large and complex. It has, in fact, defied attempts at full-length crystallization. However, efforts have been successful at biophysical characterizations of exon 1. Exon 1 encodes a 17 residue HttNT sequence followed by the variable number of polyQ repeats and a proline-rich region (polyP). The importance of exon 1 in Huntington’s disease was brought out in early studies showing that expression of exon 1 alone was sufficient to recapitulate most of the features of the disease. For that reason exon 1 is its most significant feature. In addition to exon 1, the N-terminal Htt domain contains (1) a largely unstructured region subject to cleavage by proteolytic enzymes, and (2) multiple residues subject to posttranslational modifications. The C-terminal region does not appear to be as interesting as the N-terminal domain, but does contain nuclear import and export sequences. Lastly, Huntingtin possesses several HEAT repeat regions. Two of these are situated in the N-terminal domain and one in the C-terminal domain. All of these structural features are now discussed in greater detail.
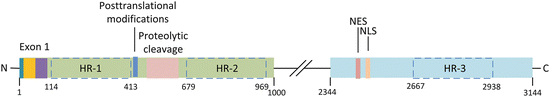
Fig. 10.2
Huntingtin structure. Exon 1 contains an N-terminal fragment (blue), PolyQ repeats (yellow), and proline-rich sequence (purple). The central portion of the N-terminal domain contains amino acid residues subject to posttranslational modifications, and a region where proteolytic cleavage by caspases and calpains occurs. The C-terminal region contains a nuclear export sequence (NES) and a nuclear localization (import) sequence (NLS). Huntingtin also has three large regions each containing several HEAT repeats (HRs); these are outlined by dashed boxes
10.2.1 The PolyQ Repeats
A landmark in understanding the significance of the glutamine repeats in HD and other polyQ disorder s was the introduction in 1994 by Max Perutz of the polar zipper concept. In Perutz’s polar zipper, the polyQ chains form β-sheets held together by hydrogen bonds between main-chain and side-chain amides. These bonds form the links of the zipper. In introducing his model, Perutz had observed that (1) polyQ tracts readily bind other polyQ tracts and form insoluble clumps, and (2) many transcription factors contain polyQ tracts and these sequences may serve a normal role in transcription. Htt with its polyQ tract of regularly spaced glutamines is able to bind not only to itself but also to these other polyQ containing proteins. In HD, the Htt polyQ stretches misfold into β-sheets that oligomerize and form fibrillar structures highly resistant to degradation and clearance by the ubiquitin–proteasome system. This basic idea has been built upon and expanded in the intervening years.
10.2.2 The PolyQ Flanking Sequences
As shown in Fig. 10.2 the polyQ repeats located in exon-1 are accompanied by N- and C-terminal flanking sequences. There is a 17-residue sequence N-terminal to the polyQ repeats (HttNT) and a 38-residue proline-rich sequence C-terminal to the polyQ’s. The latter consists of three components—two strings of proline residues separated by a proline-rich stretch. In general, both HttNT and polyP influence the folding, aggregation and toxicity of the polyQ repeats. The extent of their influences varies depending on several factors including the polyQ tract length, modifications to the sequences, and cellular context; in some cases, the influences are profound, in others they are negligible. The flanking sequences and their varying influences are discussed in greater detail in the next section.
10.2.3 Proteolytic Cleavage and Aberrant Splicing
One of the most consistent features of the polyQ disorder s is the presence of fragments of the mutated proteins in the neuronal intranuclear inclusions. These fragments are generated by cleavage of the full-length proteins by members of the caspase and calpain families of proteases. Evidence for this type of processing has been found for huntingtin, the androgen receptor, atrophin-1, ataxin-3, and ataxin-7. The huntingtin protein possesses a number of caspase and calpain cleavage sites. These cleavage operations generate N-terminal fragments that are toxic to the striatal neurons. As shown in Fig. 10.2 these sites are clustered together in a region in between the two N-terminal heat repeat regions (from residues 469 to 586).
Cleavage by calpains is significant for yet another reason. Calpains are Ca2+-dependent cysteine proteases. When activated these proteases target critical signaling elements, thereby facilitating synaptic remodeling and memory formation. Under normal operating conditions calpain activity is maintained at a low, controlled level, but Ca2+ overload triggers calpain hyperactivation as part of a protective feedback response aimed at substrates such as NMDA receptors that enable calcium entry.
Lastly, aberrant splicing of exon 1 has been found to lead to the formation of small Htt fragments. Small exon-1/intron-1 polyadenylated mRNA transcripts have been detected in brains of mice expressing mtHtt. Translation of these transcripts produces the exon-1 Htt fragments. It was surmised in that study that associations of splicing factors such as SFSR6 with the expanded polyQ tracts precipitate the aberrant splicing events.
10.2.4 Posttranslational Modifications
Huntingtin is subject to different kinds of posttranslational modification—phosphorylation, acetylation, palmitoylation, ubiquitination, and SUMOylation. These actions couple cellular signaling and regulatory pathways to the protein so that huntingtin’s subcellular localization, clearance, and processing are responsive to cellular needs. One cluster of sites where phosphorylation and acetylation occurs is located just outside the first HEAT region. Those sites are shown in Fig. 10.2. Other sites are situated further C-terminal to that region in between the HEAT repeat sequence s .
A particularly interesting locus for posttranslational modifications is situated in the HttNT. That short stretch contains phosphorylation sites at serine 13 and serine 16 and competitive ubiquitination—SUMOylation sites at lysine 6, lysine 9, and lysine 15. Ubiquitination usually targets the protein or protein fragments for proteasomal clearance, while SUMOylation stabilizes the protein and reduces aggregation. Concerted actions by the enzymes responsible for phosphorylation and ubiquitination/SUMOylation take place with phosphorylation preparing the adjacent sites for subsequent Ub/SUMO modifications. By these and similar actions, posttranslational modifications influence huntingtin’s stability, clearance, subcellular localization, aggregation, fibrillization, and toxicity.
10.2.5 The HEAT Repeats
These structures serve as platforms or scaffolds that enable multiple proteins to come into close contact with one another, thereby mediating their interaction and signaling roles. The acronym HEAT stands for huntingtin, elongation factor 3, protein phosphatase 2A, and TOR1 protein kinase. These are the first four proteins found to contain these stacked structures. HEAT repeats are amino acid sequences approximately 47 or so residues in length. Each repeat is composed of a helix-turn-helix motif; these are stacked one after the other to form curved rod-like structures, hence the name α-RODs. These structures expose large amounts of solvent-exposed surface for binding of large molecules hence their suitability as mediators of protein–protein and protein–DNA interactions.
10.3 Role of Exon 1 in Huntingtin Aggregation
The biophysical properties of the expanded polyQ repeats depend on all three components of exon 1. That is, they depend on HttNT, the number of polyQ repeats, and the polyP. Those dependences are illustrated in the next two figures. The first of these figures (Fig. 10.3a) illustrates the kinetic acceleration capability of the HttNT, while the polyP sequence slows the progression and serves in a protective capacity. The second figure (Fig. 10.3b) shows the dramatic speedup in aggregation as the polyQ length increases. The data included in these figures was obtained at several different micromolar concentrations and those values are indicated in the plots.
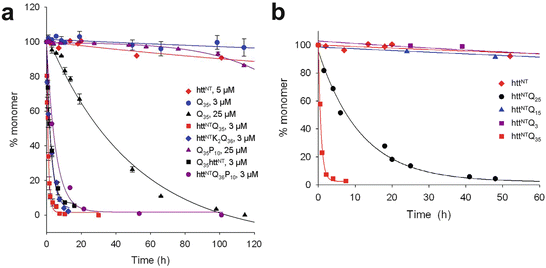
Fig. 10.3
Huntingtin exon 1 aggregation kinetics. (a) Data showing the effects of appending HttNT and P10 peptides to Q35 and Q36 repeats. (b) Illustration of the role of polyQ repeat length on the aggregation kinetics (from Thakur Nat. Struct. Mol. Biol. 16: 380 © 2009 Reprinted by permission from Macmillan Publishers Ltd)
The HttNT, by itself, tends to adopt an α-helical conformation and aggregate into α-helical oligomers. Adding the polyQ repeats to the HttNT generates a HttNT-polyQ peptide. This structure first assembles into oligomers via N-terminal helix-helix interactions and then converts to a form in which polyQ beta-hairpins or other beta-stranded structures occupy a nascent core and the N-terminal alpha helices reside on the periphery. The importance of the N-terminal segment in physiological settings is enhanced by its membrane-binding ability. The N-terminal α-helical segment anchors the Htt fragments to lipid membranes and by that means facilitates the formation of close associations between fragments. These aggregation-promoting activities are enhanced by the presence of acidic phospholipids in the membranes.
The proline-rich region situated C-terminal to the polyQ repeats has a role, too. As can be seen in Fig. 10.3a the addition of a P10 stretch reduces the rate of monomer depletion and inhibits aggregation. It does so by shifting the monomer equilibrium towards aggregation-incompetent conformations. It and other, longer stretches interfere with the formation of the beta sheets and shift the ensemble equilibrium towards formation of more compact, helical structures. They destabilize non-fibrillar aggregates and oligomers and by that means reduce their toxicity.
Overall, the HttNT and polyP segment act synergistically to:
Decrease the overall solubility of polyQ-bearing fragments,
Destabilize non-fibrillar aggregates and intermediates, and
Accelerate fibril formation.
10.4 Htt Misfolding, Oligomerization, and Aggregation
10.4.1 Aggregates and Intranuclear Inclusions
Like other disease-causing proteins such as Aβ and α-synuclein, Htt oligomers of different sizes and morphologies may be produced. The ensemble of forms includes besides monomers, native and misfolded, small annular structures and large ring-shaped ones, and still larger amorphous aggregates and well-developed fibrillar structures. The intranuclear inclusions that sit at the top of the hierarchy of forms consist of granules, filaments, and randomly and parallel-oriented fibrils.
Proteins other than Htt and fragments thereof are often present in these inclusions. One prominent group consists of proteins associated with the maintenance of proteostasis in the cell—molecular chaperones and components of the ubiquitin–proteasome system. Another important group of proteins present in the NIIs are transcription factors, especially those possessing polyQ stretches. Lastly, the amyloid-like fibrils are composed primarily of N-terminal fragments of varying sizes.
In huntingtin, there is evidence in support of both models of fibril formation. First, there is good evidence for a classical nucleated polymerization mechanism of fibril formation. In this pathway, homogeneous monomers or tetramers misfold into conformations with a high beta-sheet content. The monomers and small oligomeric species serve as seeds for the rapid, polymerization phase of fibril growth that follow the slow, lag-phase dominated nucleation step. Secondly, studies have been made of heterogeneous mixes of conformers with varying propensities to form fibrils. Results of theoretical studies of these entities support the alternative picture in which oligomers first form and these then undergo a nucleated conformational conversion of their constituents into fibril-forming beta-sheet conformers.
10.4.2 Action of Molecular Chaperones
Recall from Chap. 5 that some chaperones refold partially unfolded and misfolded proteins; others maintain solubility and prevent aggregation, or re-solubilize clumped proteins (Fig. 5.7). Still other molecular chaperones, especially those activated under conditions of chronic stress, direct protein aggregates to the UPS and/or into autophagic pathways for disposal (Fig. 5.10). PolyQ-containing aggregates, and especially those containing mutated, cleaved and modified htt, are difficult to handle. These are either targeted for degradation by the UPS or autophagy, or alternatively, sequestered in intracellular inclusion s .
Interestingly, many of the neurodegenerative-disease-causing mutated proteins including α-synuclein (PD), the androgen receptor (SMBA), and tau (FTLD) are Hsp90 “clients”; that is, they are bound and stabilized by the molecular chaperone. That has led to the development of Hsp90 inhibitors. These are intended to prevent the stabilization of aberrant proteins by Hsp90, thereby promoting their clearance by the UPS, autophagy, and sequestration .
10.5 Clearance of mHtt by the UPS, Autophagy and Sequestration in Inclusions
10.5.1 The UPS and Autophagy
Mutant Htt and its proteolytic fragments are cleared by the proteasome, but imperfectly. The proteasome cannot entirely digest the chains. Instead, it releases a variety of fragments for further hydrolysis. The fragments, if not cleared, can aggregate and further promote the onset of disease. In addition, there is a jamming issue in which excessively long polyQ peptides impair the UPS resulting in failed degradation of other misfolded and aggregated proteins. These actions precede inclusion body formation, and give rise to the idea that intracellular inclusion body formation is, in fact, a protective cellular response, and indicative of stressed protein quality control .
Autophagy takes up much of the burden of clearing full-length and fragmented mHtt. Recall from Chap. 6 that Ulk1, a key initiator of autophagy, is bound to the mTOR signaling complex. Htt aggregates interact with the mTOR complex and with Ulk1. These interactions free Ulk1 from mTOR inhibition and enable it to initiate autophagy. Htt also interacts with p62. It seems that unlike other aggregation- and neurodegeneration-prone proteins, huntingtin has a normal role as a regulator of autophagy. As a first step, members of the HSPB and DNAJ families of molecular chaperones bind hydrophobic patches in the HttNT region of huntingtin fragments that have been released from the proteasome. This action disrupts their further aggregation and fibrillization. However, the beneficial interactions between Htt and the autophagic machinery are either too weak or go awry in mutant-driven disease-situations. Empty autophagosomes have been observed in which cargo engulfment has failed to occur, and defects in autophagosome transport have been uncovered in which lysosomal membrane fusion and degradation have not taken place.
10.5.2 Sequestration in Inclusions
The TCP1 ring complex/chaperonin containing TCP1 (TRiC/CCT) chaperonins were briefly discussed in Chap. 5. Recall from that discussion that these are Type II chaperonins belonging to the Hsp60 family. These molecular chaperones complete the folding of difficult-to-handle and kinetically trapped proteins began by Hsp70 family members. TCP1, a subunit of CCT, interacts with mHtt. Through this interaction CCT promotes the incorporation of mHtt into large inclusion bodies . Once there, they are bound by Hsp70, and that action prevents cell death pathways from being activated. These sequential actions serve to detoxify neurons expressing mHtt by sequestering them in the largely inert inclusions.
A number of proteins are sequestered in cytoplasmic and nuclear aggregates by mutant huntingtin. Heat shock protein s and transcription factors were already mentioned. One of the proteins involved in gene transcription that is bound by mHtt is the CREB binding protein, CBP, a transcription co-activator. This protein, consistent with Perutz’s seminal 1994 observation, contains a polyglutamine stretch. However, the sequestration of proteins by mHtt extends beyond interactions with polyQ-bearing proteins belonging to the transcription machinery. For example, the polyP sequence in exon 1 is capable of mediating the sequestration of proteins containing WW and SH3 domains. Those interactions are implicated in the sequestration of several vesicle associated proteins by truncated wtHtt and by mHtt fragments. Thus, protection by sequestration has its limits, reached when too many proteins that are needed are tied up in these structures.
It is widely believed that the proteostasis network consisting of molecular chaperones, the ubiquitin–proteasome system, endoplasmic reticulum associated protein degradation, and the three kinds of autophagy decline with age. That decline, coupled to the continued and increasing presence of misfolded proteins that require clearance, eventually tips the scale in favor of a buildup of toxic species. Mutant huntingtin and other neurodegenerative, disorder-associated misfolded proteins exacerbate this shifting balance by tying up declining supplies of key proteostasis components and sequestering them in intracellular inclusion s . Consistent with this picture, ubiquitin, Hsp40 family members, and other components of the proteostasis network are found attached to NIIs containing N-terminal fragments of mHtt. This scenario provides a plausible explanation for the late-onset age-dependence of HD, PD, AD, and other neurodegenerative disorders.
10.6 Huntingtin Membrane Localization and Trafficking Roles
Huntingtin full-length and N-terminal fragments associate with lipid membranes. The protein–surface interactions are important for several reasons. They (1) influence the subcellular localization of normal and mutant forms of these proteins, (2) promote aggregation, and (3) help determine the toxicity of the oligomers. It is a partnership. Among the physicochemical factor contributed by the oligomers are its flexibility, hydrophobic surface exposure and stability, while the surface provides lipid composition, especially cholesterol and acidic ganglioside GM1 content, along with physical properties such as charge, membrane curvature and lipid phase. Together, these physicochemical factors determine which lipid membranes are used by Htt in carrying out its normal functions, and the relative toxicity of their abnormal offspring at those locations or at alternative ones.
Huntingtin is found in multiple membrane-associated locations. It resides most often in the cytosol but sometimes in the nucleus. Huntingtin is seen at the plasma membrane, found connected to synaptic vesicles, and tied to microtubules. It localizes to mitochondria, the endoplasmic reticulum, and the Golgi complex. Huntingtin has a role in endocytic and exocytic vesicular trafficking , and, as discussed above, is a component of the autophagic machinery.
Recall from Chap. 6 that microtubule-mediated transport is used to move cargo over long distances, while actin filaments mediate transport locally over short distances. In fast axonal transport (FAT), cargo is transported along microtubule tracks at rates up to 1 μ/s. Slow axonal transport takes place at speeds 100 times lower, on the order of 1 mm/day. Mitochondria are transported by FAT, but at reduced rates punctuated by stops and reverses designed to deliver mitochondria where they are needed.
Huntingtin mediates long-distance transport. As depicted in Fig. 10.4 Htt interacts with Huntingtin-associated proteins HAP-1 and Hap-40, with dynactin, and with regulators of vesicle endocytosis such as Rab5. By those means Htt serves as a scaffold for the attachment of cargo to the microtubule-based kinesin and myosin motor protein s . However, mutant huntingtin and especially mutant N-terminal fragments perturb the scaffolding operation resulting in stalled movement and impaired axonal transport of not only mitochondria and other organelles but also signaling proteins such as brain-derived neurotrophic factor (BDNF). It does so, at least in part, by binding and titrating HAP-1, dynamin (needed for endocytosis) and other vesicle-associated proteins in inclusion bodies . In addition, mutant huntingtin affects mitochondrial fusion–fission dynamics. It binds the mitochondrial fission GTPase DRP1, thereby disrupting fusion–fission dynamics and further preventing the normal anterograde and retrograde movement to and from synapses. There is also evidence that mHtt cause excessive S-nitrosylation of DRP1 leading to mitochondrial fragmentation and synaptic damage. The importance of S-nitrosylation in neurological disorders was discussed in the context of PD in Chap. 9.
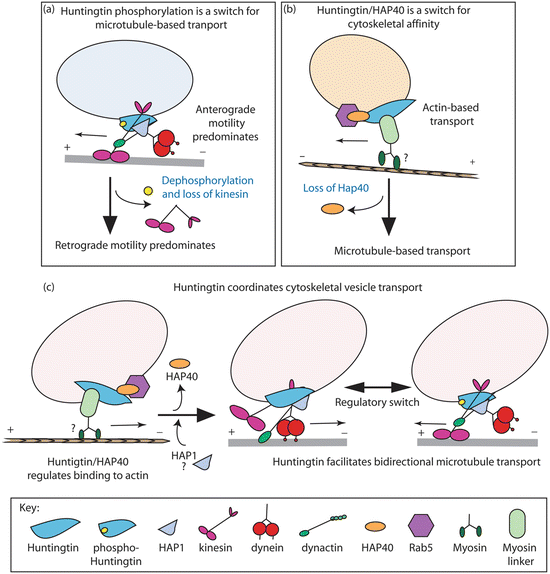
Fig. 10.4
Facilitation of vesicular transport by huntingtin. (a) Phosphorylation at serine 421 serves as a molecular switch between anterograde (kinesin) transport and retrograde (dynein/dynactin) motility. (b) Htt/Hap40/Rab5 complex mediates endosome mobility between microtubules and actin filaments. (c) Coordination of cytoskeletal vesicle transport by huntingtin, which mediates attachment to actin and bidirectional transport along microtubules. In all these models, huntingtin functions as a scaffolding protein (from Caviston Trends Cell Biol. 19: 147 © 2009 Reprinted by permission from Elsevier)
10.7 Intracellular Vesicular Trafficking: BDNF and TrkB
Growth factor signaling pathways are utilized not only during development but also in adult situations. Brain derived neurotrophic factor (BDNF) is synthesized in cortical cells. It is then transported down the axons and secreted out across the corticostriatal synaptic cleft where it binds to TrkB and p75NTR receptors on striatal dendrites. The ligand–receptor complexes are subsequently internalized, undergoing retrograde vesicular transport to the cell body where they trigger pro-survival responses that maintain the health of the striatal neurons. Lack of proper BDNF-stimulated signaling has been implicated in several psychiatric disorders including depression, addiction, schizophrenia, and the childhood disorder known as Rhett syndrome.
The TrkB and p75NTR receptors act in opposition to one another. TrkB, a high-affinity BDNF receptor, activates a number of transcription factors that support neural survival and continued synaptic plasticity . In contrast, signaling via p75NTR, a low-affinity BDNF receptor, negatively regulates synaptic plasticity and dendritic spike morphology. Under normal conditions there is a careful balance between these actions. That balance is disturbed by mutant huntingtin. This is accomplished in two major ways. First, wild-type huntingtin acts at the transcriptional level to promote transcription of BDNF. It accomplishes this by sequestering an inhibitory element called REST/NRSF (repressor element-1 transcription factor/neuron restrictive silencer factor) in the cytoplasm, thereby freeing the BDNF promoter from this inhibitory complex. Mutant huntingtin is defective in this action resulting in a BDNF deficit.
Secondly, mutant huntingtin acts at the transport level in both presynaptic and postsynaptic neurons to disrupt the TrkB/p75NTR balance. It carries out this second set of actions by inhibiting BDNF and TrkB transport. In its role as scaffolding protein wild-type huntingtin facilitates transport of BDNF along microtubules. As noted in the preceding section, mutant huntingtin, in contrast, impedes proper movement along the rail system . In addition to its facilitation of anterograde transport of BDNF ligands, huntingtin mediates retrograde transport of TrkB receptors. Here again, mutant huntingtin fails to perform its normal functions and, instead, reduces the efficiency of the transport system.
10.8 At the Synapse: Htt and NMDA Receptors
N-Methyl-d-aspartate (NMDA) receptors are the primary CNS receptors for the excitatory neurotransmitter glutamate. To recap, these receptors are dual voltage and neurotransmitter gated. Membrane depolarization relives an Mg2+ channel block, thereby enabling subsequent glutamate binding to open the receptor channel and enable Ca2+ ions to enter the neuron. Activation of these receptors initiates a host of protective effects in the host neurons through Ca2+ mediation of synaptic plasticity , gene transcription, and pro-survival activity (Fig. 10.5). Paradoxically, strong activation of these receptors has deleterious effects though Ca2+ driven anti-survival, apoptotic, and gene transcription actions. The term excitotoxicity was coined almost a half a century ago by James Olney to describe the toxic effects exerted throughout the brain by excessive calcium entry resulting from glutamatergic hyperactivation of NMDA receptors. The deleterious effects of excessive calcium entry was discussed with regard to both Alzheimer’s disease and Parkinson’s disease in the preceding chapters.
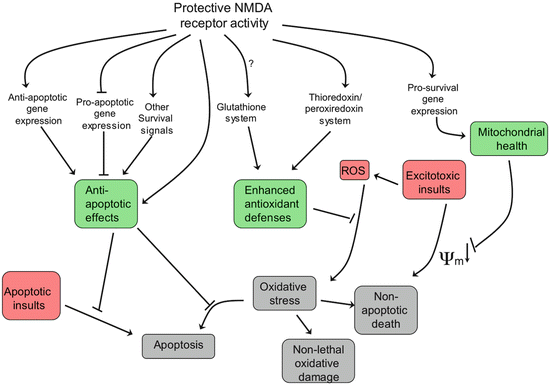
Fig. 10.5
Health-promoting responses to NMDA receptor activation (from Hardingham Nat. Rev. Neurol. 11: 682 © 2010 Reprinted by permission from Macmillan Publishers Ltd)
Recall from Chap. 8 that there are two distinct sets of NMDA receptors—one set at synapses and the other at extrasynaptic sites. The former set acts in a protective, anti-apoptotic capacity, while the latter supports cell death. The differences between the two derive from their activation of opposing cellular signaling pathways and alternate gene expression programs. Both of these regulatory routes are influenced by mHtt. Synaptic NMDA receptors activate TRiC, which by promoting mHtt aggregation prevents the more dangerous monomeric and small oligomer forms from inhibiting CRE/CREB/PGC-1α gene expression (discussed further in the next section). The extrasynaptic NMDA receptors, in contrast, act through a different agent, the small GTPase known as Ras homolog enriched in striatum (Rhes). The Rhes GTPase does the opposite. It deaggregates mHtt clusters, thereby releasing mHtt monomers and oligomers to inhibit the expression of the protective genes.
10.9 Impairment of Mitochondrial Biogenesis by mHtt
The importance of maintaining mitochondrial health was stressed in the earlier discussions of Parkinson’s disease. This theme, in fact, has become a central one in discussions of neurodegeneration, and Huntington’s disease is no exception. To see why this is so, consider for a moment brain energetics. The brain accounts for just 2 % of the body’s mass yet it consumes 20 % of the oxygen supplied to the resting body. Most of this energy is used for synaptic transmission. It is needed primarily to operate the ion pumps (chiefly Na+/K+-ATPases and Ca2+-ATPases) that restore Na+, K+, and Ca2+ ion concentrations to their resting values and repolarize the membranes following action potential generation and neurotransmitter release at the presynaptic terminal, and NMDA/other ion channel openings on the postsynaptic side. The numbers tell the story. For example, during action potential generation approximately 1 × 109 Na+ ions enter the neuron requiring the hydrolysis of roughly 4 × 108 ATP molecules to pump them out. For a mean action potential rate of 4 Hz, a total of 3 × 109 ATP molecules per neuron per second need to be supplied by the neuronal metabolic machinery, mostly through oxidative phosphorylation.
To meet this requirement mitochondria are transported and positioned in active presynaptic and postsynaptic terminals. Mitochondrial localization at active synapses is orchestrated by the transient Ca2+ influx associated with NMDA receptor activity and voltage-gated calcium channel (VGCC) openings. These signals are sensed by Miro, a mitochondrial GTPase situated in the OMM. Miro possesses a pair of EF hands, modular calcium-binding helix-loop-helix motifs. The arrival of Ca2+ ions at Miro is a stop signal and once received Miro working together with another protein, Milton, arrest movement at active axonal and dendritic terminals, thereby enabling the mitochondria to supply energy and maintain calcium homeostasis.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


