Chapter 42 Hypnotic Medications
Mechanisms of Action and Pharmacologic Effects
Abstract
Introduction
Currently, the clinically-used sedative/hypnotics in the United States include five benzodiazepines (Valium-like compounds) available in both proprietary and generic forms, three nonbenzodiazepine gamma-aminobutyric acid (GABA) agonists (zolpidem, zaleplon, and eszopiclone), and a melatonin receptor agonist (ramelteon) available as prescription agents (Table 42-1). This is a large market, involving approximately 49 million prescriptions in 2006, a growth of 53% over the previous 5 years.1 Sales in 2006 were approximately $3.7 billion. During the same year, roughly the same number of prescriptions was written for nonhypnotics prescribed for the purpose of aiding sleep; the largest among these was the antidepressant trazodone.
Table 42-1 Pharmacokinetic Properties and Dosages of Some Hypnotic Drugs Used in the Treatment of Insomnia
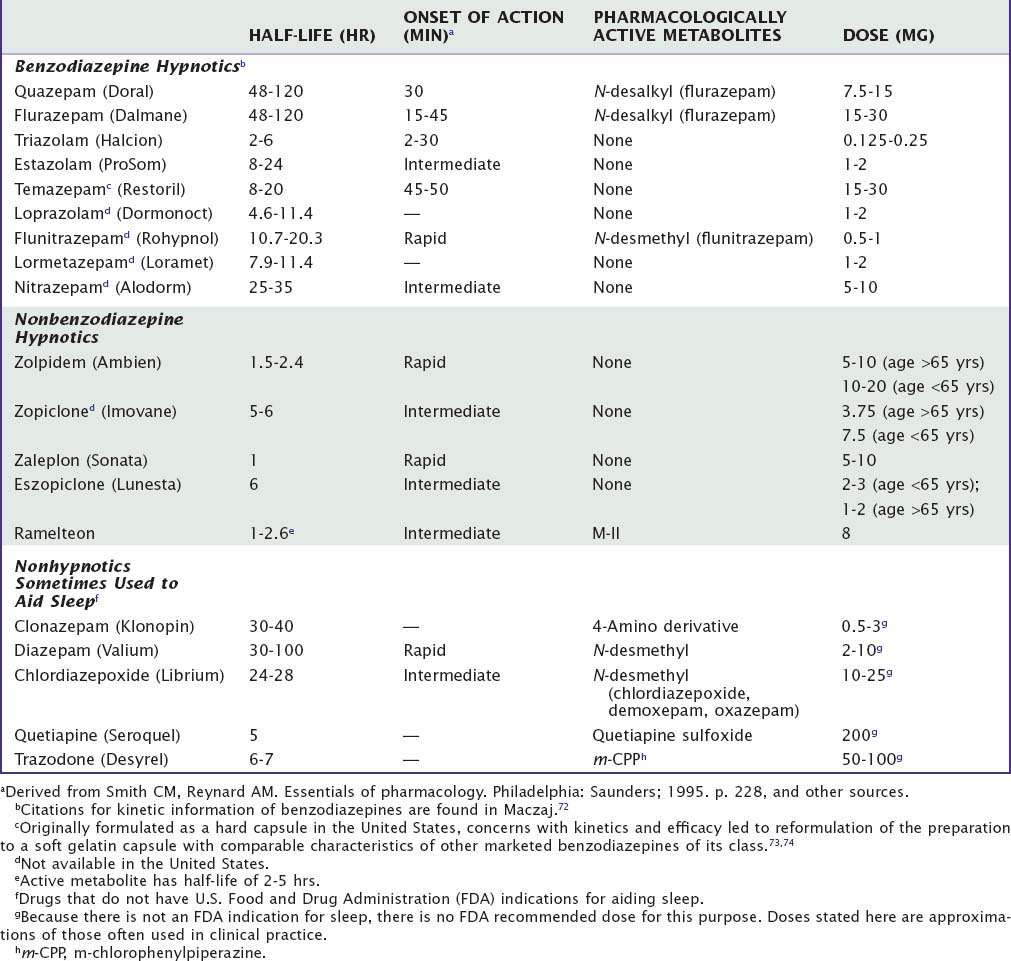
Although these are large numbers, one could make a case that in the population at large, prescription hypnotics are taken relatively infrequently by insomniacs. In a survey of over 7000 enrollees in five large health maintenance organizations,2 patients with insomnia were divided up into those with only sleep complaints (Level 1) and those who believed that their sleep difficulties had an adverse effect on their daytime functioning (Level 2). The percentage of Level 1 insomniacs taking prescription and nonprescription hypnotics was 5.5% and 11.2%; comparable values for Level 2 insomniacs were 11.6% and 21.4%, respectively. Many patients, of course, receive other classes of prescription drugs given for purposes of helping sleep. One study of primary care patients in a health maintenance organization indicated that 13% of insomniacs who were not considered to have affective disorder were receiving antidepressant medications.3 Many persons also self-medicate with alcohol; a telephone survey of approximately 1000 representative adults in the general population found that 10% had done so in the past year.4 A survey in the Detroit area reported that during the past year, 13% had used alcohol to aid sleep, whereas 18% used medication (either prescription or over-the-counter), and 5% had used both.5 Thus, alcohol use to help sleep is common even though it exposes the patient to the risk of ethanol dependence, and it is also relatively ineffective. Although orally administered ethanol has some sleep-inducing properties, it often promotes sleep disturbance as the night progresses.6
Mechanism of Action
GABAa Agonists: Benzodiazepines and the Newer Nonbenzodiazepines
When the benzodiazepines first gained prominence in the 1960s, most theories of how they might act were inherited from previous studies of ethanol and barbiturates, and involved possible alterations in membrane phospholipid integrity and energy metabolism. With the discovery of the benzodiazepine receptor (now generally referred to as the GABAA–benzodiazepine receptor complex) in the late 1970s, attention was focused on the possibility that the pharmacologic effects of these compounds result from their saturable, stereospecific binding to a specific recognition site.7,8 The receptor complex is part of a group of ligand-gated ion channel complexes that also includes the glycine, serotonin-3, neuronal nicotinic acetylcholine, and other receptors.9 It is functionally comprised of three moieties, a benzodiazepine recognition site, a GABAA recognition site, and a chloride ionophore. Molecular cloning studies indicate that structurally it is comprised of at least five different glycoprotein subunits (Fig. 42-1). The actual binding site for benzodiazepines appears to be located at the junction of the alpha and gamma subunits. Each of the subunits may have multiple isoforms.10 There appear to be as many as six alpha, three beta, and three gamma isoforms. Studies using “knock-in” technology in mice indicate that the alpha-1 subunit may mediate both sedation and memory effects of agonists.11 This suggests that it will be unlikely to develop a hypnotic that acts at this site and that will have sleep-promoting, but not amnesic, properties.
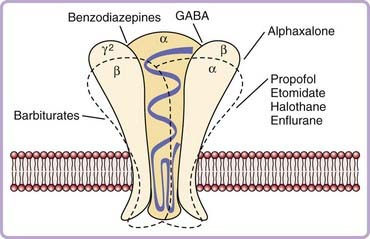
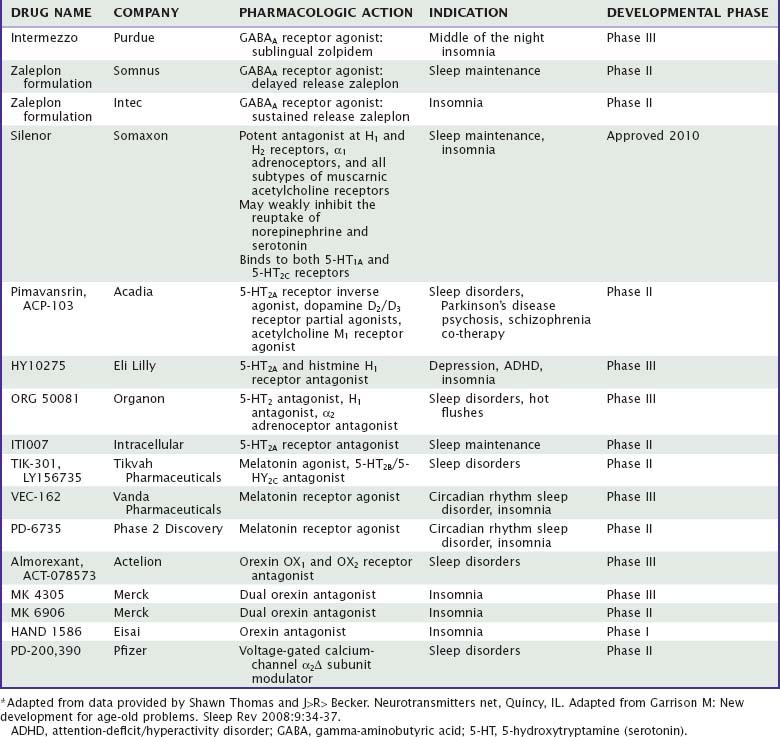
The end result of a benzodiazepine agonist binding to its specific site is that as an outcome of a complex interaction with the GABA recognition site, the flow of negatively charged chloride ions into the neuron is enhanced, which changes the postsynaptic membrane potential and alters input resistance such that the postsynaptic neuron is less likely to achieve an action potential. This inhibitory mechanism is a very potent one in the central nervous system (CNS), because the GABAA receptor is the most widespread receptor mechanism in inhibitory synapses, comprising up to 30 percent of all synapses in the CNS. One of the intriguing features of this view is that it provides a beginning for understanding a long-standing puzzle—how hypnotic medications of many different pharmacologic classes may have relatively similar effects on the process of inducing sleep. As mentioned previously, the newer nonbenzodiazepine agents such as zolpidem and zaleplon bind to a subclass of benzodiazepine recognition sites, ethanol has profound effects on chloride channel function,6 and barbiturates bind to a distinct site.12 Barbiturates, for instance, may cause chloride channels to open for prolonged periods,13 whereas benzodiazepines may increase the frequency of opening.14 Similarly, the active metabolite of chloral hydrate9 and the anesthetic propofol15 modulate GABAA-receptor function. Several lines of evidence have also suggested that the hypnotic effects of benzodiazepines may involve presynaptic effects mediated by alterations in potential-dependent calcium channel activity.16 The calcium channel blocker nifedipine, for instance, can block the sleep-inducing effects of microinjections of triazolam into the medial preoptic area. Currently, at least two hypnotics under development address the function of auxiliary alpha-2 delta subunits of voltage-gated calcium ion channels (Table 42-2).
The original characterization of the receptor complex indicated that it mediates the anxiolytic, muscle relaxant and anticonvulsant effects of benzodiazepines.7,8 That it also mediates the sleep-inducing effects was demonstrated in a series of studies showing, for instance, that some beta-carboline compounds that act as receptor inverse agonists induce increased wakefulness, and at low doses they block sleep induction by benzodiazepines.16 The binding of benzodiazepines is stereospecific, such that an enantiomeric form (the B10 compounds) have opposite effects, one induces sleep whereas the other promotes wakefulness.6,16
Neuroanatomic Considerations
In contrast to the growing understanding of the interaction of benzodiazepine agonists at a molecular level, the anatomic sites at which they act to induce sleep has been more poorly understood. The most parsimonious view would be that hypnotics act at loci thought to be important in sleep regulation on the basis of lesion or stimulation studies (a general review of the neuroanatomy of sleep is found in Chapter 7). Using that approach, Mendelson17 microinjected triazolam into a number of such sites. One of the most striking findings was how in many areas injections produced no effect on sleep (e.g., the locus coeruleus, the gigantocellular tegmental fields, the basomedial nucleus of the amygdala, and the ventrolateral preoptic area), or actually enhanced wakefulness (the dorsal raphe nuclei). In contrast, injections into the medial preoptic area (MPA) of the anterior hypothalamus consistently enhanced sleep, an anatomically very specific effect insofar as injections into nearby structures (the lateral preoptic area, the horizontal limb of the diagonal band of Broca) had no effect. The basal forebrain and anterior hypothalamus have long been thought to have an important role in sleep regulation. The MPA is a complex structure that receives afferents from many areas in the forebrain and brainstem. Among these are projections from various areas of the hypothalamus, as well as serotonergic fibers from the dorsal raphe nuclei and noradrenergic projections.18 Cell bodies and fibers of different parts cross react in immunohistochemical studies with a number of neuromodulators such as Substance P and Neuropeptide Y.19 Push-pull cannula studies have reported release of catecholamines, GABA, and glutamate.20 GABA is uniformly distributed throughout the hypothalamus, and its synthetic enzyme is found in high concentrations in the preoptic area.21 GABAA–benzodiazepine receptors appear in significant concentrations here,22 suggesting that benzodiazepines might inhibit neuronal activity. The preoptic area also contains cells that are responsive to temperature, osmolarity, glucose, and steroids,23 and it receives afferents from various sensory systems.18 The basal forebrain contains neurons that increase firing during NREM (non–rapid eye movement) sleep compared to waking (“sleep-active neurons”), although higher concentrations are found in the lateral preoptic area and diagonal band of Broca’s area.24 There are also neurons that selectively increase or decrease firing rates during REM (rapid eye movement) sleep and NREM sleep.25 Thus the preoptic area in general, and the MPA in particular, appear to have a role in coordinating various systems involved in reproductive and homeostatic functions.26 Given the bidirectional interactions of sleep with cardiovascular, thermoregulatory, endocrine, and sensory systems,27 it seems likely that the preoptic area might be involved in coordinating sleep with other systems. It may be, then, that the preoptic area is among the loci at which benzodiazepine agonists act to induce sleep. Recent work has also indicated that microinjections of pentobarbital and other GABA receptor agonists into a brainstem area, the mesopontine tegmental anesthesia area, induced an anesthesia-like state, raising the possibility that it may be involved in drug-induced loss of consciousness.28
Other Neurotransmitters
Subsequent studies have also indicated that microinjections of pentobarbital,29 ethanol,30 adenosine,31 and propofol32 into the MPA enhance sleep. Indeed, looking at a somewhat broader area, adenosine has been shown to accumulate extracellularly in the basal forebrain cholinergic region,33 which in turn has efferents to cortical and thalamic systems involved in arousal; it has been suggested that alteration of functioning of this cholinergic area may be the mechanism by which the propensity to sleep is enhanced by prolonged wakefulness. Similarly, it may be that possible some of the sleep-promoting effects of antihistamines and tricyclic antidepressants (many of which have significant anticholinergic properties) might be mediated by alterations of cholinergic neurons in the basal forebrain. The histaminergic system, which is centered in the tuberomammillary nucleus of the lateral hypothalamus, sends efferents to the preoptic area34 as well as the perifornicular area35,36 and the cortex, so it also seems possible that sedative effects of antihistamines and some tricyclic antidepressants might be mediated by altering the influence of the tuberomammillary nuclei on these centers. It has also been observed that microinjection of the GABA receptor antagonist gabazine into the tuberomammillary nuclei inhibits the effects of centrally administered GABA-ergic agents, suggesting that this area may be involved in their pharmacologic actions.37
There has been increasing interest in the hypocretin/orexin system, which is centered in the perifornicular region of the posterior and lateral hypothalamus, as a mechanism of arousal. There also is evidence that various defects in this system are associated with the genesis of narcolepsy (see Chapter 84). Microinjection of triazolam into this area shortens sleep latency and increases total sleep time in rats, raising the possibility that such hypnotics reduce wakefulness, either by direct action at the peri-fornicular area or indirectly via GABAergic outputs from the preoptic area.38 In summary, the manner in which hypnotics may function to induce sleep is not fully elucidated, but a number of findings suggest that the basal forebrain and anterior hypothalamus will be among the crucial areas to consider.
Clinical Effects
Moving from the neuroanatomic to the clinical level, less is understood about the mechanism by which hypnotics act. One approach raises the possibility that, among their actions, hypnotics alter the perception of sleep and wakefulness.39 This notion grew out of the classical observation by Rechtschaffen40 that poor sleepers, when experimentally awakened early in stage 2 sleep, tend to report that they had been awake, while good sleepers tend to report that they had been asleep. Later studies by Mendelson41,42 replicated this observation and demonstrated that after administration of triazolam, flurazepam, or zolpidem insomniacs were more likely to report that they believed they had been asleep compared to when they were given placebo. In contrast, when flurazepam or zolpidem were given to normal subjects, this effect was not evident.43 One interpretation of these data is that hypnotics such as triazolam and zolpidem may correct a misperception of sleep in some insomniacs, such that their experience of whether they are awake or asleep becomes more like that of good sleepers.
Other Sleep-Inducing Agents
Most over-the-counter hypnotics are first-generation antihistamines including diphenhydramine and doxylamine. Although they possess to varying degrees other properties including anticholinergic effects, their common quality is inhibition of the histamine-1 receptor. (Later generations of antihistamines are more hydrophilic, and in principle enter the CNS less readily, thus producing less sedation.) Tolerance to daytime sleepiness appears to develop rapidly, in about 4 days.44 As discussed earlier, antihistamines may produce drowsiness by inhibiting the histaminergic pathways centered in the tuberomammillary nucleus of the posterior hypothalamus, which enhance wakefulness. It should also be noted that some tricyclic antidepressants, notably doxepin, which at the time of this writing is under development as a possible hypnotic, have significant antihistaminic (H1 and H2) properties (although also having a number of other effects, including antagonism at alpha1 adrenoreceptors and muscarinic cholinergic receptors, and binding to serotonin 2a and 2c receptor subtypes).
Although there is debate about the effectiveness and range of side effects of melatonin as a hypnotic,45 there is now a potent agonist of melatonin subtypes I and II receptors available (ramelteon), and others in development (agomelatine, TIK-301, VEC-162). It has been hypothesized that melatonin and agonists might act to affect circadian systems via effects on melatonin receptors in the suprachiasmatic nucleus, but the ways in which it might alter sleep have not been well understood. It has been suggested that binding of agonists to the melatonin type I receptor decreases the waking signal from the suprachiasmatic nucleus. One possibility is that sleep-promoting effects of agonists at melatonin receptors are at least in part GABAergic. Melatonin administration is known to raise GABA concentrations in the rat hypothalamus, as well as 3H-diazepam binding in the forebrain.46 Similarly, decreases in motor activity produced by melatonin in the hamster are prevented by the benzodiazepine receptor blocker flumazenil.47 The author’s laboratory has reported that microinjections of melatonin into the medial preoptic area of the rat hypothalamus enhance sleep, suggesting that its site of action may be similar to that of benzodiazepines, barbiturates, adenosine, and ethanol, as described previously.48
Although the topic of anesthetics is beyond the scope of this chapter, we will briefly mention the intravenous agent propofol, which has made it much more practicable to induce anesthesia for prolonged periods in, for instance, intensive care unit settings. Neurochemically, its mechanism of action has not been fully elucidated, though it is known to interact with the GABAA–benzodiazepine receptor complex, with resultant decreases in acetylcholine release from the frontal cortex and hippocampus, and to also increase functional activity of dopamine and serotonin in the cortex. Interestingly, microinjection of propofol into the medial preoptic area of rats induces sleep, and this effect is blocked by the benzodiazepine receptor blocker flumazenil.49

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


