Benign Childhood Epilepsy with Centrotemporal Spikes
Although Martinus Rulandus described the first case of rolandic epilepsy in 1597 (
17), its specific electrographic and clinical features were recognized only during the past 50 years. In 1952, Gastaut (
18) noted that these “pre-rolandic” spikes were unrelated to focal pathology, and 2 years later, Gibbs and coworkers (
19) observed that the discharges may occur without clinical seizures. In 1958, Nayrac and Beaussart (
20) described the clinical symptoms in 21 patients. The excellent prognosis of this syndrome, compared with the poor outcome in psychomotor epilepsy, was apparent in the early literature (
21,
22).
Although BCECTS is easily recognized in its “pure” form, atypical features are common and may make a confident diagnosis difficult.
Genetics
The hallmark centrotemporal sharp waves are often found in siblings of children with BCECTS. An autosomal dominant inheritance with an age-specific expression has been suggested (
34,
35,
36), but most children with these EEG features never experience clinical seizures, which suggests that development of BCECTS depends on other genetic and environmental factors (
37,
38,
39,
40). The first positive evidence for linkage was found on chromosome 15q14, and either the α
7 acetylcholine receptor subunit gene or another closely linked gene may be implicated in pedigrees with BCECTS, but the disorder is genetically heterogeneous (
41).
Scheffer and coworkers (
42) described a syndrome of autosomal dominant rolandic epilepsy with speech dyspraxia that may also present an opportunity to identify genes involved in BCECTS. This syndrome begins at a mean age of 5.3 years, and its electroclinical manifestations are identical to those of BCECTS, with nocturnal rolandic seizures and centrotemporal spikes with a horizontal dipole. Oromotor and speech dyspraxia are also present, however, and epilepsy and cognitive impairment worsen in subsequent generations.
Clinical Manifestations
Seizures characteristically occur either shortly after falling asleep or before awakening. However, in 15% of patients they happen both in sleep and wakefulness, and in 20% to 30% of patients in the waking state alone (
31,
32). During wakefulness, simple partial seizures are the rule, with (a) unilateral paresthesias of the tongue, lips, gum, and cheek; (b) unilateral clonic or tonic activity involving the face, lips, and tongue; (c) dysarthria; and (d) drooling (
21). Stiffness of the jaw or tongue and a choking sensation are also common. Three types of nocturnal seizures have been described: (a) brief hemifacial seizures with speech arrest and drooling in a conscious state (simple partial seizures); (b) hemifacial seizures, but with loss of consciousness (complex partial seizures), often with gurgling or grunting noises that may terminate with vomiting; and (c) secondarily generalized convulsions (
32). Postictal confusion and amnesia are rare (
47).
In a small number of cases, Loiseau and Beaussart (
48) noted unusual paresthesias or jerking of a single arm or leg, abdominal pain, blindness, or vertigo, which likely reflected seizure foci outside the centrotemporal region, as children with BCECTS may also have interictal sharp waves in other areas. Very young children with BCECTS commonly present with hemiconvulsions instead of the typical facial seizure (
32). Rarely, partial motor seizures may change sides without becoming generalized (
32).
Seizures are usually infrequent. Loiseau and coworkers (
49) reported that 35 (20.8%) of 168 patients suffered a single seizure, and only 10 (5.9%) had very frequent events. Kramer and colleagues (
50) described only 13 of 100 patients suffering a single seizure. Very frequent seizures may be more likely in children whose epilepsy begins before 3 years of age (
50). Seizures often occur in clusters, followed by long seizure-free intervals. There is no known correlation between severity of the EEG abnormality, seizure frequency, and final outcome (
32).
Postictal Todd paresis occurs in 7% to 16% of cases and often suggests a focal onset in patients who present with an apparently primarily generalized seizure (
48,
51,
52).
In most children, seizures last from seconds to several minutes; however, status epilepticus has been described in some children with atypical features (
51,
52). Temporary oromotor and speech disturbances may be associated with intermittent facial twitching, a disorder that resembles anterior opercular syndrome and correlates with very frequent or continuous spike discharge in the perisylvian region (
53,
54,
55,
56,
57). This problem does not always respond to intravenous antiepileptic medication, and long-term use of steroids has sometimes seemed beneficial (
53). Eventually, all children recover over 6 months to 8 years. Some are left with mild speech disfluency or minor slowing of tongue or jaw movements, while others are completely normal. Positron emission tomography demonstrated a bilateral increase of glucose metabolism in the opercular regions in one patient with this type of nonconvulsive status (
58). Recognition of this unusual presentation as a benign focal epilepsy is important to avoid epilepsy surgery before spontaneous resolution occurs at a later age.
A few children whose initial presentation is typical for BCECTS evolve to “atypical benign partial epilepsy” or
“pseudo-Lennox” syndrome (
59,
60,
61,
62,
63). In addition to partial motor seizures, they develop frequent atonic, atypical absence and myoclonic seizures, often with nonconvulsive status epilepticus, as well as cognitive and behavioral disturbances. The sleep electroencephalograms show nearly continuous, bilaterally synchronous spike-and-wave activity. Although these children ultimately have remission of their epilepsy, many are left with varying degrees of mental handicap.
Children with BCECTS usually have an uneventful medical history, although 6% to 10% experience neonatal difficulties (including 3% with neonatal seizures) and 4% to 5% have preceding mild head injuries (
32,
40). Up to 16% have antecedent febrile seizures, which are likely to be focal or prolonged (
40).
The incidence of migraine may be increased in children with BCECTS, although studies have lacked uniformity in migraine diagnosis (
64,
65). Giroud and colleagues (
64) compared four groups of children and found migraine in 62% of 42 children with BCECTS, 34% of 28 children with absence epilepsy, 8% of 38 children with partial epilepsy, and 6% of 30 children with head trauma. Bladin (
65) followed up 30 cases of BCECTS, noting that 20 (67%) patients had recurrent headaches during the evolution of seizures and 24 (80%) developed typical migraine after remission of BCECTS. The pathophysiologic link between BCECTS and migraine is unknown.
Electroencephalographic Manifestations
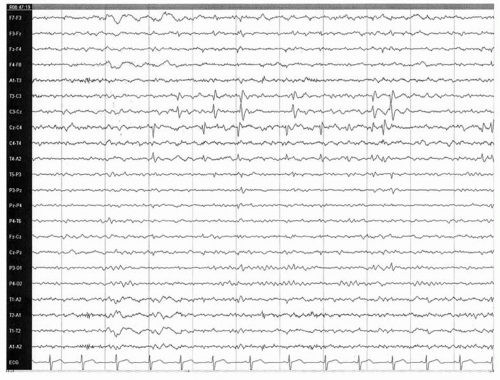
The electroencephalogram shows high-amplitude diphasic spikes or sharp waves with a prominent aftercoming slow wave (
Fig. 24.1). Spikes have a characteristic horizontal dipole, with maximal negativity in centrotemporal (inferior rolandic) and positivity in frontal regions (
66,
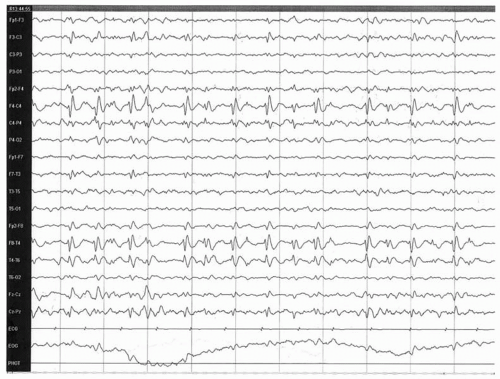
67). They frequently cluster and are markedly activated in drowsiness and nonrapid eye movement sleep (
Fig. 24.2); approximately 30% of patients show spikes only during sleep (
68). The focus is unilateral in 60% of cases and bilateral in 40%, and may be synchronous or asynchronous (
32). The location is usually centrotemporal. Legarda and coworkers (
69) described two electroclinical subgroups of BCECTS: a
high-central group, with maximum electronegativity at C
3/C
4 and seizures with frequent hand involvement, and a low-central group, with maximum electronegativity at C
5/C
6, ictal drooling, and oromotor involvement.
Atypical locations are frequent, and spikes may shift location on subsequent EEG recordings. On 24-hour electroencephalograms in children with typical BCECTS, Drury and Beydoun (
70) found 21% of the patients to have a single focus outside the centrotemporal area. Follow-up recordings showed shifts in foci both toward and away from the centrotemporal area; no clinical differences were found between patients with foci in or outside the centrotemporal zone. Only half of their patients had a horizontal dipole. Wirrell and coworkers (
51) also noted atypical spike location in 17% of their patients. Mild background slowing has been observed (
47,
51), and while generalized spike-and-wave discharge may be present, coexistent absence seizures are rare. With remission, spikes disappear first from the waking record and later from the sleep recording (
32).
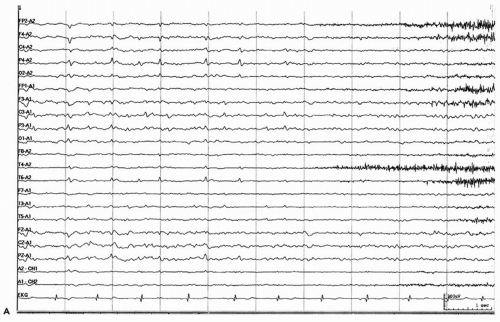
Few reports of recorded rolandic seizures (
Fig. 24.3) exist, but two unique features are noted (
32,
71,
72,
73,
74,
75). Ictal spike-and-wave discharges may show dipole reversal, with electropositivity in the centrotemporal region and negativity in the frontal area; postictal slowing is not seen.
BCECTS develops in only a small minority of children with typical rolandic spikes or sharp waves on their electroencephalograms. Rolandic discharge was found in 27 of 3726 (0.7%) waking EEG recordings of normal children without a history of seizures (
37); if recordings had included sleep, the actual number might have been higher. The percentage of children with rolandic sharp waves who develop clinically apparent seizures is unclear. On the basis of reported incidences of BCECTS and of rolandic EEG discharge in normal children, the risk is probably less than 10%. Therefore, rolandic discharges should be considered most likely an incidental finding in children with other potentially nonepileptic presentations such as staring spells.
Neuropsychological Aspects
A variety of neuropsychological problems have been identified in children with active epilepsy and EEG discharge.
Several early studies noted frequent behavior problems, hyperactivity, inattention, and learning disorders in children with BCECTS, but attributed these difficulties to the social stigma of epilepsy or to side effects of antiepileptic medication (
33,
49,
76). More recent comparisons with normal controls yielded interesting results. On neuropsychological assessments, Croona and colleagues (
77) found that children with BCECTS had more problems than matched controls with auditory-verbal memory, learning, and some executive functions, as evidenced by poorer performance on the trailmaking, verbal fluency, and Tower of London tests. No deficits were documented in immediate memory or visuospatial memory and learning. Teachers noted greater difficulty with reading comprehension in the BCECTS group, but not to the degree predicted by the neuropsychological data. In other comparisons with controls, Gunduz and coworkers (
78) described more problems in go-no-go testing and language and minor motor deficits, and Baglietto and colleagues (
79) documented poorer performance on tests of visuospatial short-term memory, attention, cognitive flexibility, picture naming, verbal fluency, and visuoperceptual and visuomotor coordination. Other uncontrolled studies (
80,
81) have reported high rates of learning disorders and difficulties with impulsiveness and temperament in children with BCECTS.


 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access




