Idiopathic Generalized Epilepsy Syndromes of Childhood and Adolescence
Tobias Loddenkemper
Selim R. Benbadis
Jose M. Serratosa
Samuel F. Berkovic
TERMINOLOGY
The term idiopathic generalized epilepsy (IGE) was defined in the International Classification of Epilepsies and Epileptic Syndromes in 1989 (1). In addition to establishing the difference between partial and generalized, this classification added the terms idiopathic, cryptogenic, and symptomatic. In the previous version of the classification, IGEs were referred to as “primary” generalized epilepsies and the symptomatic and cryptogenic types as “secondary” generalized epilepsies (2). However, confusion existed, because secondary in the context of a generalized epilepsy meant “secondary to cause,” whereas in the context of generalized seizures it referred to seizures with focal electroencephalographic (EEG) features followed by generalization.
In all other areas of medicine idiopathic usually means “of unknown cause”; IGEs, however, actually describe a genetically low threshold for seizures. This unusual meaning was not changed in the most recent multiaxial classification (3), in which an idiopathic epilepsy syndrome is defined as “a syndrome that is only epilepsy, with no underlying structural brain lesion or other neurologic signs or symptoms. These are presumed to be genetic and are usually age dependent.” IGE syndromes are characterized by onset in childhood, good response to medical treatment and good prognosis, lack of a structural cerebral abnormality, and a presumed genetic cause (4). The absence of an underlying brain abnormality may be debatable, as more evidence for focal structural (5,6), functional (7), and biochemical (8) heterogeneity and abnormalities becomes available.
Specific Idiopathic Generalized Epilepsy Syndromes as a Continuum
Specific syndromes in the current classification of IGEs are childhood absence epilepsy (CAE); juvenile myoclonic epilepsy (JME); juvenile absence epilepsy (JAE); benign myoclonic epilepsy in infancy; epilepsy with generalized tonic-clonic seizures only; epilepsy with myoclonic astatic seizures (Doose syndrome); epilepsy with myoclonic absences; and generalized epilepsies with febrile seizures plus. However, separation of specific IGE syndromes may be difficult, and many patients may be placed in unspecified categories. To determine the frequency of epilepsy syndromes, pediatric patients with IGE were studied as a cohort: 76% received a diagnosis “idiopathic generalized epilepsy, not otherwise specified” (9), a classification that could not be changed even after 2 years of follow-up. Many authors therefore refer to IGE as a spectrum of disorders rather than as multiple separate entities (10, 11, 12, 13, 14). A study at a tertiary epilepsy center found 12 of 89 randomly selected patients to have IGE, but only 4 of these 12 could be placed into a specific subcategory of IGEs (15). Further evidence for overlap among the spectrum of IGE derives from genetic studies suggesting that the analysis of seizure
types as compared to IGE syndromes may be more helpful in genetic linkage studies (16).
types as compared to IGE syndromes may be more helpful in genetic linkage studies (16).
Response to pharmacologic treatment and prognosis is usually good. Benbadis and coworkers reviewed the initial treatment regimen of 58 patients with EEG-confirmed IGE (17). Only 29% were on adequate pharmacologic treatment whereas 48% were treated with ill-advised antiepileptic drugs (AEDs) only and 22% on a combination of ill-advised and adequate AEDs (17). Therefore, improvement in recognition of IGE and appropriate choice of medication is crucial to management and outcome in these patients.
GENERALIZED IDIOPATHIC EPILEPSY SYNDROMES OF CHILDHOOD AND ADOLESCENCE
Juvenile Myoclonic Epilepsy
History
Sudden and brisk muscular contractions during epileptic seizures were first described in the second century by Galen (18). The term myoclonus is derived from Friedreich’s description of paramyoclonus multiplex to indicate symmetric and multifocal jerks (19). Other 19th century authors, such as Delasiauve (20), Gowers (21), Féré (22), and Reynolds (23), also mentioned the isolated myoclonic jerks that occur in some epilepsy patients, but it was Herpin (24), in 1867, who gave the first detailed description of a patient with JME. He called the myoclonic jerks “secousses,” “impulsions,” and “commotions épileptiques,” that is, compulsions that shake the body as an electric shock would.
In Penfield and Jasper’s Epilepsy and the Functional Anatomy of the Human Brain, Penfield used the term “myoclonic petit mal” (25). In 1957, Janz and Christian described the characteristics of the syndrome in 47 patients (26). The following year, Castells and Mendilaharsu independently described 70 patients with “bilateral and conscious myoclonic epilepsy” (27). Jeavons, in 1977, was the first to describe the excellent response of patients with JME to valproate (28,29). In 1984, Asconape and Penry (30) and Delgado-Escueta and Enrile-Bacsal (31) reported the clinical and electrophysiologic characteristics of this syndrome.
Epidemiologic Factors
JME is estimated to account for approximately 10% of all epilepsies, with a range from 4% to 11% (12,30,32, 33, 34, 35). Underdiagnosed in the past, JME recently became a well-recognized epilepsy syndrome. Previously, myoclonias were frequently recognized only after detailed history taking or after recording myoclonic seizures on video-electroencephalography. The attention given to genetic studies and the widespread use of video-EEG monitoring have made JME an easily recognizable syndrome. Its true prevalence in the general population is difficult to ascertain, as many patients may misinterpret myoclonic seizures as clumsiness or nervousness (36). There is no male or female predominance in most series.
Clinical Features
Age of Onset
According to Janz, JME begins between 12 and 18 years of age (mean, 14.6 years) (32,37) but can manifest in all age groups (38). Dialeptic (or absence) seizures may occur earlier (mean, 11.5 years; range, 8 to 16 years) than myoclonic seizures (mean, 15.4 years; range, 8 to 30 years) or generalized tonic-clonic seizures (mean, 15.5 years; range, 8 to 30 years) (39).
Precipitating Factors
Myoclonic seizures and generalized tonic-clonic seizures occur mainly on awakening in the morning (40) or after afternoon naps (30) and are triggered by sleep deprivation, fatigue (41), and alcohol (42). In 41% to 42%, menstruation may precipitate myoclonic seizures (30,31). Cocaine and marijuana appear to aggravate seizures in JME (43).
Neurologic Examination
Imaging
Imaging studies do not reveal a cause for the epilepsy. Quantitative magnetic resonance imaging has shown that patients with JME, as well as those with other IGEs, have a significantly larger volume of cortical gray matter than control subjects, with frequent abnormalities in the regional distribution of cerebral gray and white matter (5,6). In JME patients, this cerebral abnormality affects the mesial frontal lobes more severely. Swartz and colleagues (44), using 18F-2-fluoro-2-deoxyglucose positron emission tomography during visual working memory tasks, found differences in regional metabolic patterns between JME patients and control subjects. Their findings support the concept that individuals with JME may suffer from cortical disorganization and abnormal patterns of cortical activation that are associated with subtle cognitive dysfunction.
Seizure Semiology
Myoclonic Seizures
The typical myoclonic seizures of JME consist of mild to moderate jerks of the neck, shoulders, arms, or legs. The jerks are more frequent in the upper than in the lower extremities and are usually bilaterally symmetric but may
be unilateral (12,31,45,46). Lancman and associates (45) found asymmetric myoclonic seizures manifested as predominantly unilateral or initially unilateral jerks followed by bilateral jerks in 11 of 85 patients with JME. Myoclonic jerks of the upper extremities can often cause patients to drop objects and can interfere with morning activities such as eating breakfast, brushing the teeth, or applying cosmetics. The jerks can be single or repetitive and frequently involve extensor muscles. If repetitive, they are not rhythmic and usually do not exceed more than a few movements. Falling to the floor is uncommon, but falls may occur when patients are in an awkward position and are “surprised” by a myoclonic seizure. The amplitude of the movement is variable, but the jerk is not forceful or massive. Recovery is immediate, without loss of consciousness. These myoclonias should be distinguished from the massive sudden myoclonic seizures that propel patients to the ground with great force (as seen in Lennox-Gastaut syndrome and in progressive myoclonus epilepsies). A series of repetitive myoclonic jerks may precede a generalized tonic-clonic seizure, producing a clonic-tonic-clonic seizure (47).
be unilateral (12,31,45,46). Lancman and associates (45) found asymmetric myoclonic seizures manifested as predominantly unilateral or initially unilateral jerks followed by bilateral jerks in 11 of 85 patients with JME. Myoclonic jerks of the upper extremities can often cause patients to drop objects and can interfere with morning activities such as eating breakfast, brushing the teeth, or applying cosmetics. The jerks can be single or repetitive and frequently involve extensor muscles. If repetitive, they are not rhythmic and usually do not exceed more than a few movements. Falling to the floor is uncommon, but falls may occur when patients are in an awkward position and are “surprised” by a myoclonic seizure. The amplitude of the movement is variable, but the jerk is not forceful or massive. Recovery is immediate, without loss of consciousness. These myoclonias should be distinguished from the massive sudden myoclonic seizures that propel patients to the ground with great force (as seen in Lennox-Gastaut syndrome and in progressive myoclonus epilepsies). A series of repetitive myoclonic jerks may precede a generalized tonic-clonic seizure, producing a clonic-tonic-clonic seizure (47).
Sometimes the myoclonic seizures of JME are perceived only as an electric shock or a “feeling” inside the body, and there is no evidence of movement. Consciousness is either not affected or is so briefly and mildly impaired that the deficit is difficult to perceive. This clearly distinguishes the myoclonic jerks of JME from dialeptic seizures associated with myoclonic jerks (“myoclonic absences”) in which shoulder, arm, and leg jerks occur against a background of loss of consciousness, during which patients are unaware of the seizure as jerking continues (48). In contrast, just after a myoclonic seizure, JME patients know that a seizure has occurred. Even successive jerks may not impair consciousness (12).
Generalized Tonic-Clonic Seizures
Generalized tonic-clonic seizures are often preceded by a few minutes of generalized, mild to moderate jerks of increasing frequency and intensity, a seizure pattern known as clonic-tonic-clonic. Because consciousness is preserved during myoclonic jerks, patients may predict a grand mal seizure a few minutes before it occurs and adopt a safe position to prevent major injuries. Consciousness is abruptly lost when the head, face, neck, and trunk extend with a tonic contraction. The tonic phase lasts approximately 10 to 20 seconds and leads to a final phase of clonic trunk and limb jerks. Generalized tonic-clonic seizures are present in 87% to 95% of patients with JME (33,37,49) and are frequently precipitated by lack of sleep, alcohol intake, and stress (41,42).
Dialeptic Seizures (Absences)
Typical dialeptic seizures (or absences) occur in 10% to 33% of JME patients (31,37,49,50). Dialeptic seizures of JME are relatively infrequent, of short duration, and not associated with automatisms (51,52). A prospective study in JME found dialeptic seizures in 16 (31.9%) of 42 patients (53). Clinical changes were subtle and became apparent only with adequate testing. When dialeptic seizures occurred before the age of 10 years, the patient would stop activities, not answer questions, and stare. No postictal symptoms or memory recollections of ictal events were observed. When dialeptic seizures appeared after the age of 10 years, the manifestations were usually less severe and consisted of subjective instant loss of contact and concentration or brief impairment of concentration revealed on questioning (53).
Electroencephalographic Findings
Interictal Electroencephalography
The characteristic EEG pattern of JME consists of discharges of diffuse bilateral, symmetric, and synchronous 4- to 6-Hz polyspike-wave complexes (Fig. 25.1). Discharges may be accentuated over the frontocentral regions. The interictal complexes usually have two or more high-voltage (150 to 300 μV) surface-negative spikes, widespread but of higher voltage in the anterior head region (32). Delgado-Escueta and Enrile-Bascal (31) found 4- to 6-Hz polyspike-wave complexes in all untreated individuals and diffuse 3-Hz spike-wave complexes in 17% of patients. Asymmetric, as well as focal, EEG abnormalities have also been described (45). The resting electroencephalogram background is normal.
Ictal Electroencephalography
The ictal electroencephalogram demonstrates 10- to 16-Hz generalized polyspikes followed by slow waves. Also seen are 4- to 6-Hz diffuse polyspike-wave complexes.
Genetics and Etiology
In 1973, Tsuboi and Christian (54) studied families of patients with “primary generalized epilepsy and sporadic myoclonias of impulsive petit mal.” Of 319 probands, 27.3% had at least one near or distant relative affected with epilepsy. Of 390 family members, 15% had diffuse polyspike-wave complexes, and another 39.8% had nonspecific paroxysmal EEG abnormalities. Inheritance was interpreted to follow a polygenic-threshold pattern, with female relatives being more affected than male relatives.
Family studies of JME since then have resulted in conflicting results regarding the mode of inheritance. Polygenic (54), digenic (recessive-dominant or recessive-recessive) (55), single-gene autosomal dominant (56), and recessive models have been proposed. In the families of JME patients, affected family members usually express different idiopathic generalized epilepsy syndromes, such as childhood absence epilepsy, juvenile absence epilepsy, or epilepsy with generalized tonic-clonic seizures on awakening (57, 58, 59, 60). Some asymptomatic family members show
the typical 4- to 6-Hz polyspike-wave complexes on the electroencephalogram (59,61).
the typical 4- to 6-Hz polyspike-wave complexes on the electroencephalogram (59,61).
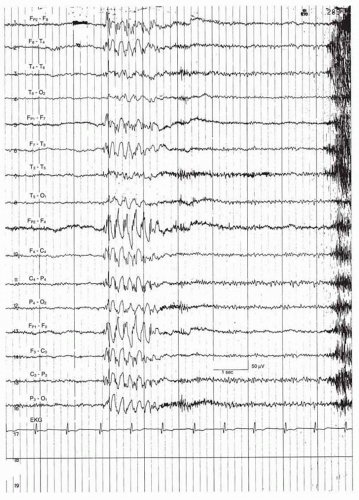 Figure 25.1 Interictal 4- to 6-Hz polyspike-wave discharge of frontocentral predominance in juvenile myoclonic epilepsy. No clinical changes were seen, and the patient could recall a word given during the discharge. |
Recent studies revealed that JME is a heterogeneous entity related to several mutations. Major genetic susceptibility loci for JME have been termed EJM 1, 2, and 3. Depending on modifying genetic mutations in other loci, phenotypic expression can vary.
In addition, there have been descriptions of individual JME families that appear to be clearly affected by a major autosomal recessive (62) or dominant gene (61). Autosomal dominant inheritance patterns have been described in association with mutations in the GABRA1 gene on chromosome 5q34-q35 (63), the CACNB4 gene on chromosome 2q22-q23 (64), and the CLCN2 gene on chromosome 3q26 (65).
The term EJM 1 is used to characterize the JME phenotype caused by EFHC1 gene mutations located on 6p12-p11. Families with JME from Mexico, Belize, and Los Angeles had high limit of detection (LOD) scores at the 6p11-p12 locus (66, 67, 68). Another study from the Netherlands confirmed the 6p12-p11 marker in conjunction with JME (69). In 2004, Suzuki and colleagues (70), demonstrated mutations in the EFHC1 gene in six different families, including
the ones previously described by Liu and associates (67,68) and Bai and coworkers (66).
the ones previously described by Liu and associates (67,68) and Bai and coworkers (66).
EJM 2 is used to characterize the mutation in the α7 subunit of the neuronal nicotinic acetylcholine receptor on chromosome 15q14. Based on the previous description of autosomal dominant nocturnal frontal lobe epilepsy as a mutation in the α4 subunit (CHRNA4) of the neuronal nicotinic acetylcholine receptor (71, 72, 73), linkage analysis of this region was performed in 34 families with JME and suggested that this region was a major susceptibility locus for JME (74). A maximum multipoint LOD score of 4.42 was obtained at a point 1.7 centimorgans (cM) telomeric to marker D15S144. In this study, 25 of 27 sib pairs had identical haplotypes by descent in one or both chromosomes in the chromosome 15 region. Haplotype sharing analysis allowed placement of the candidate gene in a 15.1-cM region on chromosome 15q. Of interest, a transgenic mouse deficient in this subunit showed generalized hypersynchronous 4- to 7-Hz sharp wave activity on EEG, similar to EEG abnormalities found in JME patients.
EJM 3 is used to characterize a mutation in the BRD2 gene, a presumed transcription regulator on chromosome 6p21(75). This locus was initially named EJM1 (76), but now mutations in the EFHC1 gene on 6p12-p11 are called EJM 1 because of phenotype variations. Greenberg and coworkers (55) performed segregation analysis on 28 pedigrees. Including asymptomatic family members with abnormal electroencephalograms as affected or unaffected, these authors rejected the fully penetrant recessive model. Initial linkage studies reported evidence of linkage to chromosome 6p using human leukocyte antigen (HLA) and properdin (Bf) as markers (59). The clinical phenotype for this 6p epilepsy locus consisted of myoclonic seizures, generalized tonic-clonic or clonic-tonic-clonic seizures, absences, and, in asymptomatic individuals, diffuse 4- to 6-Hz polyspike-wave complexes. Maximum LOD scores were obtained using a recessive mode of inheritance with 60% penetrance. Subsequently, other studies reported similar findings (58,77).
A single pedigree, including 10 members showing JME or asymptomatic polyspike-wave complexes on EEG, has supported linkage to chromosome 6p by resulting in LOD scores of 3.4 for several 6p markers (61). Recombinant events defined the JME gene region to be a 43-cM interval flanked by marker D6S258 (HLA region) and marker D6S313 (centromere). Results in this large family independently prove linkage of JME and the subclinical polyspike-wave EEG pattern to chromosome 6p markers. Another study by Sander and coworkers (78) has provided further evidence of a JME locus on chromosome 6p. This locus would confer susceptibility to idiopathic generalized seizures in a majority of German families of JME patients. In this study, the 6p gene was proposed to be in a 10-cM region on chromosome 6p between HLA-DQ and marker D6S1019.
Pal and colleagues noted a linkage disequilibrium between JME and markers in the critical region 6p21, with a peak in the area of the BRD2 gene (75). DNA-sequencing demonstrated two JME-related single nucleotide polymorphisms in the BRD2 promotor region, and could not identify any other related mutation (75). Of interest, reported abnormalities of cerebral microanatomy as reported in JME would be compatible with BRD2 involvement (6).
A mutation in the GABRA1 gene on 5q34-q35 has been described in 14 members of a French-Canadian family with JME (63). Transmission was autosomal dominant. The authors speculate that seizures are caused by a dysfunction in this ligand-gated channel, because the mutation in the channels produces a reduction in GABA-activated currents in vitro (63).
Additionally, a mutation of the CACNB4 gene on chromosome 2q22-q23 encoding for the β4 subunit of a voltage-dependent calcium channel has been described in a patient presenting with bilateral myoclonic jerks of the shoulders, rare dialeptic seizures, and generalized tonic-clonic seizures after awakening. This patient was diagnosed with JME. Of note, the proband’s daughter presented with generalized 3-Hz spike-and-wave complexes without clinical change (64).
Furthermore, a CLCN2 gene mutation encoding for a chloride channel 2 on chromosome 3q26 has been identified in a family presenting with serial myoclonic jerks, frequently followed by a generalized tonic-clonic seizure (65). Four family members were diagnosed with autosomal dominant JME and one member had epilepsy with generalized tonic-clonic seizures only (65). The mutation causes functional alteration of the channel function leading to increased chloride concentration within the cell, reduced γ-aminobutyric acid (GABA) response, and neuronal hyperexcitability and seizures (65).
Pharmacologic Treatment
Valproic acid is effective in 86% to 90% of JME patients (29, 30, 31,79, 80, 81), stopping myoclonias and tonic-clonic, clonic-tonic-clonic, and absence seizures without significant side effects. Newer AEDs, such as lamotrigine or topiramate, may also be useful, but valproic acid enjoys the widest experience. As soon as a diagnosis of JME with grand mal convulsions is entertained, treatment with valproic acid or an alternative drug should be started. Seizures invariably return after drug withdrawal, and lifetime treatment is usually required (31).
Therapeutic doses of valproic acid range from 20 to 30 mg/kg. Patients receiving monotherapy require a lower dose than do those taking valproic acid in combination with other AEDs (29). Most patients are controlled with blood levels of 40 to 100 mg/mL, but in resistant cases, levels of up to 150 mg/mL may be necessary. Penry and colleagues (81) reported that 50% of their patients relapsed at some time between 2 months and 9 years of valproic acid treatment. Relapses were a result of fatigue, drug noncompliance, stress, sleep deprivation, and alcohol consumption.
Lamotrigine is a useful alternative, especially when valproic acid alone is not effective or not well tolerated. In
some patients, however, lamotrigine must be combined with valproic acid for seizure control (82). Topiramate is effective in JME, but controlled studies are lacking (83).
some patients, however, lamotrigine must be combined with valproic acid for seizure control (82). Topiramate is effective in JME, but controlled studies are lacking (83).
Phenytoin, phenobarbital, or primidone may also be added as a second drug to valproic acid in resistant cases. When used as monotherapy, control rates are much lower than those for valproic acid. According to Janz, primidone monotherapy was the treatment of choice before valproic acid became available (37), and 74% of his patients were treated with phenobarbital, phenytoin, or primidone alone. With phenytoin monotherapy, up to 40% of patients experienced an increased frequency of myoclonic jerks (84).
Ethosuximide may be added to valproic acid in JME patients with uncontrolled absences. Clonazepam should be added in patients with uncontrolled jerks. Clonazepam alone is ineffective, controlling only the myoclonic jerks and not the grand mal seizures (33). As adjunctive therapy (with valproic acid or other first-line AEDs), it may be useful in patients with persistent myoclonic jerks. Used alone, clonazepam may suppress the jerks that herald a generalized tonic-clonic seizure and not allow patients to prepare for this type of attack. Some clinicians have favored acetazolamide monotherapy for patients who continue to have generalized tonic-clonic seizures despite trials of first-line AEDs or for patients who have difficulty tolerating valproic acid (85). According to these investigators, acetazolamide does not control myoclonus as well as valproic acid does, but it controls generalized tonic-clonic seizures and is a useful adjunct to valproic acid for resistant cases.
Vigabatrin and gabapentin are not indicated in JME because myoclonic jerks and absence seizures may increase. Carbamazepine, vigabatrin, and tiagabine (86) are associated with absence status epilepticus in patients with IGEs and should not be used in JME.
Levetiracetam has been tested in patients with IGE resistant to medical treatment. Of 10 patients with JME who responded, 2 became seizure free (87). Eight of nine patients responded to zonisamide, with five becoming seizure free (88).
Pregnancy, Juvenile Myoclonic Epilepsy, and Valproic Acid
Female patients receiving valproic acid should be advised of reports that link its use to neural tube defects. These defects occur in 0.5% to 1% of pregnancies in patients receiving valproic acid monotherapy, and in 1% to 2% of pregnancies in patients receiving valproic acid in combination with other AEDs. Supplementation with folic acid may ameliorate the risk. Withdrawal of AEDs in patients who suffer few myoclonic jerks only before pregnancy is planned and reinstatement after the first trimester may be a treatment option in selected patients after a discussion with the patient of the risks and benefits. All seizure precipitants should be avoided, and when myoclonic seizures on awakening suggest particular vulnerability to convulsive seizures, a fast-acting benzodiazepine should be administered orally or rectally (89). The effects of AEDs on birth control medications should also be discussed with the patient. For women of child-bearing age, lamotrigine or topiramate may be considered.
Outcome and Prognosis
JME carries an excellent prognosis because AED treatment controls seizures in most patients. Nevertheless, any condition that places a patient at risk for a generalized tonic-clonic seizure is not benign. Sudden loss of consciousness can lead to injuries or even death. In swimming or driving, for example, a generalized tonic-clonic seizure can have devastating effects. Recognition of this syndrome is important because of the good response to valproic acid and the high incidence of relapse after medication withdrawal. JME is one form of epilepsy in which discontinuation of pharmacotherapy cannot be recommended, even after long seizure-free periods. Delgado-Escueta and Enrile-Bacsal (31) found a 90% relapse rate after withdrawal of AEDs. In our experience, not a single patient has been seizure free after medication was withdrawn. The patient should be treated as soon as a diagnosis is made and, if possible, before a grand mal seizure occurs. According to Janz (12,32), patients who did not achieve freedom from seizures had their condition untreated 9 years longer than those who responded well to medication. Penry’s study showed that 86% of patients with JME are seizure free or well controlled with valproic acid alone or in combination with other AEDs (81).
Childhood Absence Epilepsy
History
CAE, the most widely recognized type of absence epilepsy, was described early in the 20th century as pyknolepsy by Sauer and Adie, who noted its childhood onset, multiple clustered minor attacks (pyknos, Greek for “crowded” or “dense”), and usual spontaneous resolution (90,91). Synonyms include petit mal epilepsy, true petit mal, and pyknoleptic petit mal.
Epidemiologic Factors
CAE accounts for 2% to 8% of patients with epilepsy (92, 93, 94, 95, 96); the variation depends largely on the mode and source of case collection. In Swedish children 1 to 15 years of age, Olsson found an annual incidence of JAE without diffuse brain damage in 6.3 per 100,000 children (97); the majority had CAE. The cumulative incidence of absence seizures (number of persons who will have absence seizures at some time in their lives) was 98 per 100,000 children in this Swedish study (97), but only 49 per 100,000 children in a Danish study (98). A female bias in affected children is
frequently observed (13,99, 100, 101, 102), although in some studies boys and girls were equally represented (103,104).
frequently observed (13,99, 100, 101, 102), although in some studies boys and girls were equally represented (103,104).
Clinical Features
Age of Onset
CAE characteristically begins between the ages of 4 and 10 years with absence seizures (13,105), but age of onset is not strictly limited and can vary between 2 and 10 years (peak incidence is about 5 years of age). Absences occurring before age 3 years are unusual, although an apparently typical case beginning at 6.5 months has been described (106). Approximately 10% of patients have febrile convulsions before the onset of absences (99,104,105,107). The latest age limit is also difficult to define, and classification of early adolescent cases as CAE or JAE can be somewhat arbitrary. Onset after 11 years of age with the typical pyknoleptic pattern of CAE is unusual (13).
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


