Idiopathic Myoclonic-Astatic Epilepsy of Early Childhood—Nosology Based on Electrophysiologic and Long-Term Follow-Up Study of Patients
Hirokazu Oguni*
Kitami Hayashi*
Kaoru Imai*
Makoto Funatsuka*
Masako Sakauchi*
Seigo Shirakawa*
Makiko Osawa*
*Department of Pediatrics, Tokyo Women’s Medical University, Tokyo, Japan
Introduction
The Commission on Classification and Terminology of the International League Against Epilepsy has most recently proposed benign myoclonic epilepsy in infants (BME) and myoclonic-astatic epilepsy of early childhood (MAE) as distinct subgroups of idiopathic generalized epilepsy and severe myoclonic epilepsy in infants (SME) and cryptogenic LGS (CLGS) as subgroups of generalized epileptic encephalopathy (1). Controversies, nonetheless, continue and are unresolved as to the clinical distinction between these four syndromes. The clinical phenotypes overlap and it is difficult to separate and differentiate between each disorder, especially between MAE and the other three syndromes (2,3,4,5,6,7,8,9). Historically, a number of studies have focused on groups of patients whose epilepsy starts in infancy and whose phenotype seizures consist of myoclonic or astatic attacks of idiopathic etiology. These groups of patients were distinguished from other patients with LGS or West syndromes. (10,11,12,13,14,15,16). Most authors support giving the idiopathic epilepsy subtypes in this age range a more favorable prognosis than the latter two malignant syndromes. However, these groups of patients unfortunately suffered from a relatively wide variety of seizure types whose prognosis also varied. Seizure phenotypes ranged from myoclonic seizures alone or astatic seizures alone or myoclonic seizures or astatic seizures in combination with atypical absence and generalized tonic-clonic or clonic seizures (GTCS). Prognosis ranged from very benign to relatively resistant or poor prognosis. Some authors tried to subclassify them into many subdivisions. Others tried to consider them as one syndrome with a wide range of clinical manifestations, based on the conceptual framework of genetically determined corticoreticular epilepsy (3,4,15). Although syndromic classification and the diagnostic limits that border each syndrome are important, more precise information regarding the neurophysiologic characteristics, treatment response, and long-term outcomes in the idiopathic patient group are needed for practical purposes and for further discussions on classification. Here we present the results of our clinical and EEG studies in patients with MAE extensively investigated at Tokyo Women’s Medical University. We discuss the nosology of this unique epileptic syndrome based on our own experiences (17,18,19,20,21,22,23).
Electrophysiologic Characteristics of the Main Seizure Types
The most important and distinct feature of seizures in MAE appear to be an astatic seizure or epileptic drop attack, repeatedly illustrated in references. The drop attack itself is not a necessary prerequisite for MAE in the original criteria (4). The advent of simultaneous video-EEG technology has disclosed that most epileptic drop attacks seen in clinical practice were caused by brief tonic seizures. Atonic drop attacks, which were originally thought of as a cause of epileptic drop attacks, are extremely rare (24,25). We have been studying the ictal polygraphs of myoclonic or astatic seizures to investigate the neurophysiologic background of these brief motor seizures, and identified true atonic drop attacks to be a cause of astatic seizures in children with MAE, most of whom were initially treatment resistant, but finally entered into remission (19,20,21,22,23). The number of ictal polygraphs of patients with MAE, which were available for detailed neurophysiologic examinations, amounted to 30, including video-EEG (n = 5), polygraph (n = 2) and video-polygraph (n = 23). Patients with SME, BME, and CLGS were excluded based on the criteria previously defined by ILAE. All attacks occurred intermittently and did not form clusters in the periodic fashion seen in infantile spasms. Analysis of ictal EEGs showed that all attacks corresponded to generalized spike or polyspike-and-wave complexes in contrast to those of tonic drop attacks observed in LGS (26,27,28,29).
The main seizure types of MAE were largely divided into atonic and myoclonic types; myoclonic in 16 cases, atonic with or without preceding minor myoclonia in 11 cases, and myoclonic-atonic in 3 cases. Although atonic seizures were, at times, preceded by minor myoclonic movement of the extremities, the major symptomatology was global atonia causing the patients to fall to the ground, when they were intense enough. In the ictal EEG of atonic seizures, the spike-and-wave morphology was similar, characterized by a positive–negative–deep–positive wave followed by a large negative slow wave (Fig. 12-1). Interestingly, when we compared the morphology of spike-and-wave complexes among atonic seizures of different intensity in two patients, the depth of the second positive component of spike-and-wave complexes corresponded well to the intensity of the attacks; the second component was deeper and the atonic seizures were stronger (22). The difference of the component was very clear when we compared the averaged spike-and-wave complexes between those causing atonia and those without atonia (Fig. 12-2).
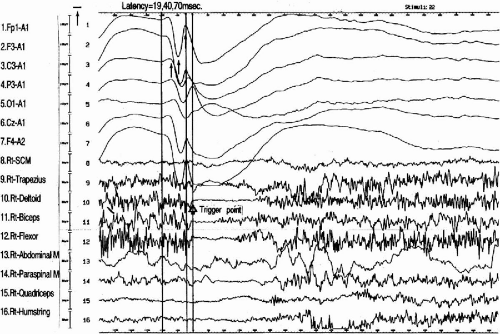 FIG. 12-1. The morphology of generalized spike-and-wave complexes causing atonic drop attacks. The polygrahic data were sampled at 1024 Hz through Rhythm Program (Stellate Inc. Montreal). Twenty-two ictal polygraphs were averaged at the onset of surface EMG interruptions of the right deltoid muscles. The latencies measured between the onset of atonia (right deltoid muscle) and each peak of the negative and positive spike component at F3 consisted of 19, 40, and 70 msec. |
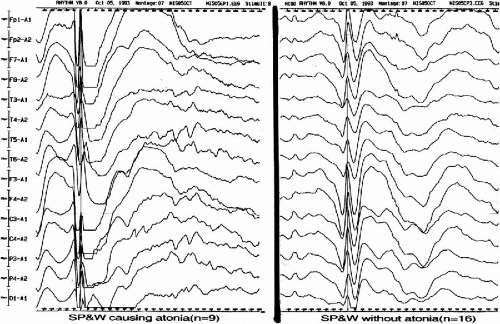 FIG. 12-2. The differences in morphology of spike-and-wave complexes with and without atonia. The morphology of the spike-and wave component was compared between that causing atonia and that without atonia in the averaged spike-and-wave complexes. Note the deep second positive component followed by large and long slow wave in the averaged spike-and-wave complexes causing atonic attack. |
The clinical manifestations of atonic seizures were characterized by a sudden collapse of the body forward, with immediate recovery, when the patients were sitting on the floor. When they were standing, their bodies and legs suddenly sagged. In more intense attacks, they collapsed straight downward onto the buttocks with an immediate recovery (Fig. 12-3A and B). Close and repeated analysis of the atonic attacks on video and surface electromyography (EMG) showed minor myoclonic jerks on the extremities preceding the atonic attacks. They were subtle enough to miss if not carefully analyzed. When patients were in the lying position, we only recognized sudden facial changes without any movement of the body or minor movement due to sudden muscle tonus reduction. Surface EMG showed sudden interruption of the ongoing EMG activity extending up to 400 msec (Fig. 12-1). Latency studies measuring onset of these EMG interruptions at various muscles in three cases with MAE demonstrated variable onsets without descending order from proximal to distal muscles (30) (Fig. 12-4). Results suggested that atonia of muscles was a secondary phenomenon, presumably due to inhibition of postural control, rather than an epileptic inhibitory impulse descending through efferent pathways, causing atonia of each of the muscles.
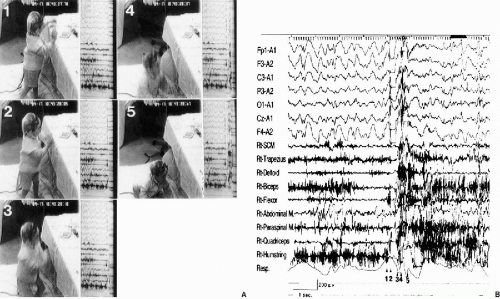 FIG. 12-3. Simultaneous video (A) and polygraphic findings (B) of an atonic drop attack, A: Before the seizure, the patient was standing in front of a desk and manipulating a toy (1). He suddenly collapsed straight downward, landing on his buttocks (2,3,4,5). Note the semiflexion at the knees (3). The arms also dropped downward (3 and 4). He had already regained consciousness by the time he landed on the floor (5). B: The EMG potentials preceding the interrupted EMG potentials were seen polygraphically on the forearm flexor and quadriceps muscles. The EMG discharges of the quadriceps and biceps femoris muscles were restored during the late phase of falling, suggesting that the bodily collapse was probably due not only to global atonia, but also to gravity, such that he immediately recovered once on the floor. Numbers in the photograph corresponded to those of the EEG. (From Oguni H, Uehara T, Imai K. Osawa M. Atonic epileptic drop attacks associated with generalized spike-and-wave complexes—video-polygraphic study in two patients. Epilepsia 1997;38:813–818. With permission.) |
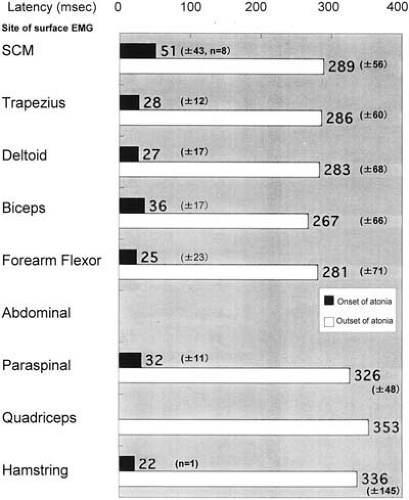 FIG. 12-4. Spreading of the atonic events among the involved muscles during atonic seizures. Latencies were measured between the peaks of the negative spike component at F3 and the onset as well as termination of the EMG interruptions in nine muscles from SCM to hamstring during the atonic drop attacks in the case shown in Fig. 12-3. Eight ictal events were individually studied to identify the efferent system responsible for the atonic seizures. The polygrahic data were sampled at 1024 Hz through Rhythm Program (Stellate Inc. Montreal). The result demonstrated atonia of muscles starting with hamstring, followed by forearm flexor, deltoid, trapezius, and finally SCM, and lasting longer in the distal than the proximal muscles. (From Tanaka T, Tsurumi E, Shirakawa S, et al. Neurophysiological analysis of the epileptic atonic drop attack. Jpn J EEG EMG (Jpn) 1998:175. With permission.) |
In the predominant myoclonic seizure type, six cases, had a history of falling down during attacks, while the remaining ten cases never had drop attacks. Among six cases with a history of drop attacks, the precise mechanism for falling was a massive propulsion or retropulsion caused by myoclonia of axial muscles in three patients. Myoclonic seizures occurred during wakefulness in ten cases, during sleep in six cases, and in the remaining one case during both sleep and wakefulness. All the attacks occurred intermittently and singly except for one child, who had the myoclonic jerk occurring twice successively. The morphology of spike-and-wave complexes during myoclonia was also similar to the morphology of spike-wave complexes during atonia. However, spike-waves of myoclonias appeared to be more brief during the duration of one complex. In one patient, drop attacks were difficult to distinguish as originating from either myoclonias or atonias when captured in the standing position, due to contamination of movement artifacts on the polygraph (Fig. 12-5). It was much easier to distinguish when they were captured in the lying position because of absence of gravity. We recorded every seizure in standing, sitting, and lying position, in order to distinguish mechanisms underlying falls. As a result, we can determine the exact seizure types underlying drop attacks or astatic attacks even by history-taking from caregivers.
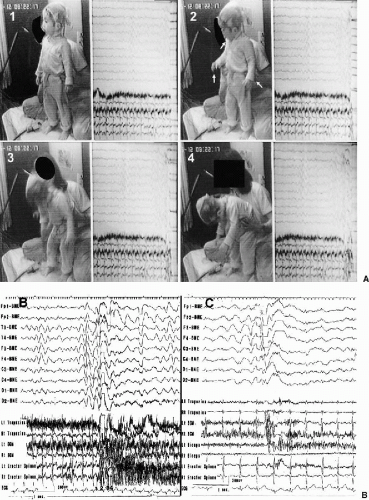 FIG. 12-5. Simultaneous video (A) and polygraphic findings (B) of the myoclonic astatic seizure A: Before the seizure, the patient was standing (1). Suddenly, slight flexion of his head as well as forearms, associated with opening of the mouth, was observed (2). Flexion of the upper trunk at the hip with abduction of both arms, as well as some flexion of both knees, ensued (3). His body was further bent forward, but he did not fall because his mother held him at the waist (4).
Related posts: Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Eyelid Myoclonia and Absence
Childhood Absence Epilepsy Evolving to Juvenile Myoclonic Epilepsy: Electroclinical and Genetic Features
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood Myoclonic Status in Nonprogressive Encephalopathies
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Eyelid Myoclonia and Absence
Childhood Absence Epilepsy Evolving to Juvenile Myoclonic Epilepsy: Electroclinical and Genetic Features
Familial Adult Myoclonic Epilepsy (FAME)
Treatment of Myoclonic Epilepsies in Infancy and Early Childhood
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|