Infections of the Nervous System
Bernard L. Maria
James F. Bale Jr.
Many different pathogens can invade and damage the developing or mature central nervous system (CNS). These infectious disorders share several clinical and pathologic features, with unique signs or symptoms attributable to the pathogen, tropism, virulence, or variations in the host immune responses. In general terms, CNS infections can be categorized according to the nature of the infectious pathogen—viral, bacterial, protozoan, or fungal—and by the location of the infection—parenchymal, meningeal, or vascular. The diagnosis of infectious disorders of the CNS requires laboratory studies, including isolation or molecular detection of the specific pathogen and the identification of organism-specific serologic or pathologic abnormalities.
MENINGITIS
Menigitis refers to inflammation of the leptomeninges, the connective tissue layers in closest proximity to the surface of the brain. Meningitis can be caused by bacteria, viruses, parasites, and fungi as well as by noninfectious conditions including inflammatory disorders (e.g., systemic lupus erythematosis or Kawasaki disease) and neoplasia (e.g., leukemic meningitis) (1). Historically, meningitis due to viruses and certain other nonbacterial pathogens has been designated “aseptic meningitis.”
Bacterial Meningitis
In the United States, approximately 25,000 cases of meningitis occur annually among children and adults (1). The pathogens causing pediatric bacterial meningitis vary considerably according to the age of the child. In newborns Streptococcus agalactiae (group B streptococcus), Escherichia coli, Staphylococcus species, Listeria monocytogenes, and Pseudomonas aeruginosa are the most frequent causes of bacterial meningitis. Citrobacter species, other potential causes of neonatal meningitis, account for less than 5% of cases but produce brain abscesses in approximately 75% of the infected infants. By contrast, two organisms, Streptococcus pneumoniae, a gram-positive diplococcus, and Neisseria meningitides, a gram-negative diplococcus, cause most cases of bacterial meningitis among children and adolescents living in regions with compulsory immunization for Haemophilus influenzae type b (Hib). Prior to the development and marketing of an effective Hib vaccine, however, as many as 1 in every 400 children between 1 and 4 years of age experienced bacterial meningitis due to this organism.
Among unimmunized children, H. influenzae meningitis remains a potential threat and occurs more commonly among African Americans and Native Americans and in children with asplenia, sickle cell anemia, and HIV infection. Risk factors for S. pneumoniae meningitis include the foregoing as well as nephritic syndrome, cochlear implantation, and cerebrospinal fluid (CSF) leaks. College students and persons with inherited complement deficiencies have increased risks of N. meningitides infection.
Pathology and Pathophysiology
Anatomic and Immunologic Features
Bacteria reach the leptomeninges by hematogenous spread, passage through the choroid plexus, rupture of superficial cortical abscesses, or contiguous spread from an adjacent infection (2). Hematogenous spread occurs during bacteremia and can result from passive transfer of organisms by infected leukocytes. Organisms can also enter the CNS from damaged blood vessels, via neurosurgical procedures, or via compound (open) fractures of the skull. In rare instances, bacterial meningitis can complicate congenital defects such as myelomeningoceles or dermal sinuses that allow direct bacterial invasion of CSF or by spread of bacteria from adjacent infections, such as parameningeal abscess, or osteomyelitis of the cranial bones.
In most instances, meningitis begins with bacterial colonization of the nasopharynx and bacteremia (2). The probability of meningitis correlates with the magnitude and duration of the bacteremia, which relate directly to the size of the intranasal inoculum. S. pneumoniae, H. influenzae, and N. meningitidis secrete proteases that neutralize
secretory IgA and impair the mucosal cilia. Invasion of N. meningitidis and S. pneumoniae across the nasopharyngeal mucosal epithelium can occur by endocytosis and transport across the cell in membrane-bound vacuoles. H. influenzae invades directly through tight junctions between columnar epithelial cells (3). Once in the bloodstream, organisms can avoid lysis by circulating complement (3).
secretory IgA and impair the mucosal cilia. Invasion of N. meningitidis and S. pneumoniae across the nasopharyngeal mucosal epithelium can occur by endocytosis and transport across the cell in membrane-bound vacuoles. H. influenzae invades directly through tight junctions between columnar epithelial cells (3). Once in the bloodstream, organisms can avoid lysis by circulating complement (3).
TABLE 7.1 Frequency of the Common Forms of Acute Meningitis in Infancy and Childhooda | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
The blood–brain barrier, composed of the arachnoid membrane, the choroid plexus epithelium, and the endothelial cells of the cerebral microvasculature, separates the intravascular compartment from the brain and CSF. This barrier effectively prevents ingress of many molecules and particles, including many infectious agents; it is unclear how most organisms causing meningitis breach this barrier (4). Pneumococci use a surface protein to bind to the receptor for platelet activating factor on cytokine-activated cerebral endothelial cells. By corrupting host chemokine receptor pathways, bacteria enter the cell, and the receptor is recycled for subsequent reuse (5). In meningitis, the blood–brain barrier can be disrupted by separation of intercellular tight junctions, primarily in the endothelial cells of the cerebral microvasculature and, to a lesser degree, in the endothelium of the choroid plexus (4,5,6). Matrix metalloproteinases, nitric oxide, reactive oxygen species, and metabolites of the arachidonic pathway participate in the disruption of the blood–brain barrier (7). Upon entering the CSF, bacteria encounter few host defenses because the CSF generally lacks antibody, complement, and opsonic activity.
Unique bacterial antigens, often capsular polysaccharides, contribute to the virulence and neurotropisms of certain bacteria (1,2,8). E. coli K1 strains cause the majority of cases of neonatal E. coli meningitis, emphasizing the importance of this antigen in the organism’s virulence. Mortality and morbidity in E. coli meningitis are greater when it is caused by K1 strains as compared with non-K1 strains. The clinical and pathologic aspects of meningitis are summarized in works by Schuchat and coworkers (9), Saez-Llorens and coworkers (10), Smith (11), and Tunkel and Scheld (12,13). Table 7.1 summarizes the frequency of several bacterial pathogens (9).
The fundamental pathologic process in bacterial meningitis is inflammation of the leptomeninges. This begins with hyperemia of the meningeal vessels, and soon thereafter, neutrophils migrate into the subarachnoid space (Fig. 7.1). The subarachnoid exudate increases rapidly and within a few hours extends into the sheaths of blood vessels and along cranial and spinal nerves. Phagocytic polymorphonuclear leukocytes predominate during the initial stages, and lymphocytes and histiocytes increase over the subsequent days. Fibrinogen and other blood proteins contribute to the inflammatory exudates, and plasma
cells appear. Fibroblasts participate in the organization of exudate and fibrosis of the arachnoid.
cells appear. Fibroblasts participate in the organization of exudate and fibrosis of the arachnoid.
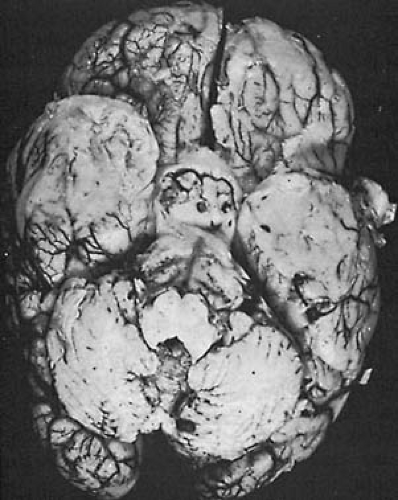 FIGURE 7.1. Haemophilus influenzae meningitis. The brain is swollen with flattening of the gyri. The meninges are thickened, and purulent material lies along the bases and over the temporal and frontal poles. (Courtesy of Dr. P. Cancella, Department of Pathology, University of California, Los Angeles, CA.) |
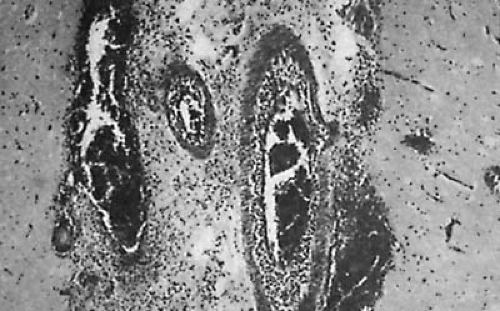 FIGURE 7.2. Haemophilus influenzae meningitis. A purulent exudate is seen in the subarachnoid space surrounding the venules, which are partially or completely thrombosed (hematoxylin and eosin stain, ×25). (Courtesy of Dr. P. Cancella, Department of Pathology, University of California, Los Angeles, CA.) |
The meningeal infection spreads along the penetrating cortical vessels in the Virchow-Robin spaces. The adventitia of these vessels, formed by an investment of the arachnoid membrane, is invariably involved, even in the initial stages of meningitis. Subarachnoid arteries also are affected. Endothelial cells swell, proliferate, and constrict the vessel lumen in 48 to 72 hours. Adventitial connective tissue becomes infiltrated by neutrophils, and a layer of inflammatory cells also appears beneath the arterial intima. Foci of necrosis of the arterial walls can develop and occasionally cause arterial thrombosis and infarction. Stroke and other vascular phenomena can be demonstrated in children with meningitis (14). A similar process occurs in the veins (15) with foci of necrosis and thrombi that can occlude the vascular lumen. Venous thrombosis is thought to occur more frequently than arterial thrombosis during bacterial meningitis (Fig. 7.2).
TABLE 7.2 Cytokines that Increase in Response to Meningitis | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||
With resolution of meningitis, cells disappear in the order in which they appeared. The number of neutrophils diminishes after a few weeks, whereas lymphocytes, plasma cells, and macrophages can persist for months. The extent of resolution depends on the stage at which infection is arrested. If infection is controlled early, residua can be minimal, but an infection lasting several weeks produces permanent fibrosis of the meninges, resulting in a cloudy arachnoid membrane and adhesions between the arachnoid and dura.
The inflammatory response elicited by bacteria and bacterial cell wall fragments contributes to meningitis-induced tissue injury (16,17). Bacterial lipopolysaccharide elicits a monocytic response in the parenchyma (18). Thus, therapy with bacterial drugs that lyse bacteria can transiently exacerbate inflammatory responses and potentially increase the severity of tissue injury. Corticosteroids, when given immediately prior to antibiotic therapy, can minimize the inflammatory response (19) and reduce the likelihood of certain complications, especially sensorineural hearing loss in children with H. influenzae meningitis.
Cytokines, soluble proteins released by host cells in response to bacterial products, participate in the pathogenesis of meningeal inflammation (20,21,22,23). Cytokines can increase blood–brain barrier permeability, decrease autoregulation of cerebral blood flow, and induce cytotoxic edema. Cytokines can recruit leukocytes into an infected compartment. Proinflammatory cytokines include tumor necrosis factor-alpha (TNF-α), interleukin (IL)-1β (IL-1β), IL-6, IL-8, IL-10, and transforming growth factor β(TGF-β) (23) (Table 7.2). TNF-α, a major contributor to the pathogenesis of bacterial meningitis and its complications, is a low-molecular-weight protein secreted by activated macrophages, leukocytes, endothelial cells, microglia, and astrocytes. TNF-α increases the permeability of the blood–brain barrier (23,34), induces cell lysis, and mediates myelin and oligodendrocyte damage (35). Levels of TNF-α are transiently elevated to more than 200 pg/mL
in CSF in the majority of patients in the first 24 to 48 hours of purulent meningitis (24). By contrast, CSF concentrations of TNF-α usually remain normal in patients with viral meningitis (36,37).
in CSF in the majority of patients in the first 24 to 48 hours of purulent meningitis (24). By contrast, CSF concentrations of TNF-α usually remain normal in patients with viral meningitis (36,37).
IL-1β, a highly potent inducer of neutrophil accumulation and procoagulant, stimulates the release of other cytokines such as TNF and IL-6 and of hypothalamic corticotrophin-releasing factor. The latter increases systemic levels of adrenocorticotrophic hormone (ACTH) (38). ACTH can increase blood–brain barrier permeability for albumin with augmentation of edema. Peak concentrations of CSF IL-1b more than 100 pg/100 mL correlate with poor outcome for neonatal gram-negative meningitis and childhood pyogenic meningitis (24,39). IL-1 and TNF-α act synergistically to disrupt the blood–brain barrier and can stimulate human vascular endothelial cells to promote transendothelial passage of neutrophils (40).
IL-6, which is also a pyrogen, plays a role in the induction and propagation of inflammatory responses. Increased levels of IL-6 are detected in CSF of patients with bacterial meningitis (30), and serum concentrations are also elevated, in contrast to the compartmentalization of the responses of other cytokines in meningitis (30,31,41). Levels of IL-8, a leukocyte chemotactic agent that promotes leukocyte adherence, are increased in CSF of patients with meningococcal meningitis but not aseptic meningitis (42). IL-10 is not present in serum but is elevated in CSF in the first 48 to 72 hours of viral meningitis. It is believed to downregulate inflammation and may contribute to chronicity of disease.
Cytokines released from damaged cells stimulate leukocyte chemotaxis from blood into CSF, a response more pronounced in the juvenile than in the adult brain (43). Chemokines include diverse agents such as platelet-activating factor, IL-b, families of cysteine-containing chemokines, and the macrophage inhibitory proteins (MIPs). Ectopic expression of chemokines in the CNS leads to dramatic accumulation of circulating leukocytes in brain parenchyma and CSF with accompanying pathology (44). Conversely, inhibition of leukocyte trafficking to the brain attenuates inflammation and tissue damage, indicating that leukocytes are an important mediator of neuronal injury. This inhibition can occur at the level of the leukocyte integrins that serve as adhesion molecules or at the level of the endothelial adhesion receptors, such as selectins and intercellular adhesion molecules (45).
Reactive oxygen and nitrogen species, potent antimicrobial factors, can also damage host cells. Microglia, when activated with interferon-beta and bacterial lipopolysaccharide, cause neuronal cell injury by a nitric oxide mechanism (46,47). Nitric oxide, a gas with a half-life of seconds, regulates vascular tone and perfusion pressure. In excess amounts, as generated in the CSF during infection, nitric oxide enhances blood–brain barrier permeability and leukocytosis (48). Administration of inhibitors of inducible nitric oxide synthase decreases the bystander damage to host cells during meningitis. The mean concentration of nitric oxide is significantly elevated in CSF during the early stages of bacterial but not aseptic meningitis (49).
When the fibrinopurulent exudate accumulates in large quantities, hydrocephalus can be produced by obstruction of the foramina of Luschka and Magendie or the aqueduct of Sylvius. Communicating hydrocephalus can result from inflammation or exudate in the subarachnoid space around brainstem and over the cerebral convexity. The exudate can interfere with CSF flow from the cisterna magna and lateral recesses to the basal cisterns or with absorption of CSF by arachnoid granules (Fig. 7.2).
Brain perfusion can be impeded by acute increases in intracranial pressure (50), changes in the vascular bed, or acute ventricular dilatation. Brain metabolism changes to glycolysis with increased glucose oxidation and lactate production and the depletion of high-energy compounds, such as phosphocreatine and adenosine triphosphate (ATP). Brain edema in bacterial meningitis can be vasogenic, the result of increased permeability of the blood–brain barrier, or cytotoxic. Both vasogenic and cytotoxic edema can be induced by components of the neutrophil membrane (51). Cytotoxic brain edema is marked by increased brain water, cellular swelling, increased intracellular sodium, and loss of intracellular potassium (51). Brain edema can depolarize neuronal membranes and lead to seizure activity. These changes are mediated in part by polyunsaturated fatty acids released from leukocytes and possibly brain cell membranes (52). Vasogenic edema, caused by the opening of tight junctions between cerebral capillary endothelial cells and increased micropinocytosis across endothelial cells, can result from the presence of bacteria as well as inflammation. Additionally, there are concomitant increases in superoxide and lipid peroxidation, believed to result from inhibition of superoxide dismutase activity. Arachidonic acid, formed in part by cleavage of phospholipids by phospholipase A2, accumulates not only in meningitis, but also in other pathologic insults such as ischemia, hypoxia, and trauma. Dexamethasone, which inhibits phospholipase A2 and stabilizes cell membranes, reduces arachidonic acid–induced brain edema in vivo but not in vitro (see Table 7.3) (52). Bacterial cells or cell fragments also induce cerebral arteriolar vasodilatation by formation of oxygen free radicals.
Gram-negative endotoxin in the CSF can induce respiratory and circulatory failure (53). Symptoms include pulmonary edema, which develops from an intense systemic vasoconstriction; subendocardial hemorrhages; myocardial necrosis; hemorrhagic lesions in the adrenal corticomedullary junction; and ulcerations of the intestinal mucosa. McCracken observed that the endotoxin concentrations in CSF correlate with clinical severity and neurologic outcome of H. influenzae type B meningitis (54).
During the early stages of meningitis, regional cerebral blood flow is increased; however, in severe meningitis cerebral blood flow is reduced (55). Paulson and colleagues observed that cerebral blood flow was reduced to 72% of normal in meningitis and the cerebral arteriovenous oxygen difference to 63% of normal, with a consequent decrease in oxygen consumption to 42% of the normal levels (56). Although autoregulation is lost in experimental meningitis, its role in the pathogenesis of childhood meningitis is less clear. Ashwal and colleagues observed that autoregulation of cerebral blood flow was preserved in children with bacterial meningitis (57). The changes in cerebral blood flow secondary to changes in carbon dioxide partial pressure (pCO2) indicate that hyperventilation can reduce cerebral blood flow below ischemic thresholds. For this reason hyperventilation to reduce intracranial pressure in meningitis may potentiate neuronal injury. Mortality in bacterial meningitis and other serious CNS infections correlates, in part, with decreased cerebral perfusion pressure (58).
During the early stages of meningitis, regional cerebral blood flow is increased; however, in severe meningitis cerebral blood flow is reduced (55). Paulson and colleagues observed that cerebral blood flow was reduced to 72% of normal in meningitis and the cerebral arteriovenous oxygen difference to 63% of normal, with a consequent decrease in oxygen consumption to 42% of the normal levels (56). Although autoregulation is lost in experimental meningitis, its role in the pathogenesis of childhood meningitis is less clear. Ashwal and colleagues observed that autoregulation of cerebral blood flow was preserved in children with bacterial meningitis (57). The changes in cerebral blood flow secondary to changes in carbon dioxide partial pressure (pCO2) indicate that hyperventilation can reduce cerebral blood flow below ischemic thresholds. For this reason hyperventilation to reduce intracranial pressure in meningitis may potentiate neuronal injury. Mortality in bacterial meningitis and other serious CNS infections correlates, in part, with decreased cerebral perfusion pressure (58).
TABLE 7.3 Therapeutic Interventions Against the Inflammatory Response | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Clinical Manifestations
The early signs of neonatal bacterial meningitis may consist only of low-grade fever, poor feeding, somnolence, “fussy” behavior, or irritability (Table 7.4). Later, vomiting, lethargy, and seizures ensue. The physical examination in infants with meningitis can be equally nonspecific, showing somnolence, irritability, hyper-reflexia, and a full or bulging fontanel. Meningeal signs are present infrequently. Systemic signs can include hypotension and features of disseminated intravascular coagulopathy (DIC).
The signs and symptoms of meningitis in older children include fever, headache, nausea and vomiting, nuchal rigidity, alterations of sensorium, convulsions, cranial
nerve palsies, disturbances in vision, and occasionally papilledema (59). Headache, often accompanied by photophobia, results from inflammation of the meningeal vessels and from increased intracranial pressure. Head retraction, neck stiffness, and spinal rigidity are caused by irritation of the meninges and spinal roots, which elicits protective reflexes intended to shorten the spinal axis and immobilize the irritated tissue (60). With lengthening of the spine, nerve roots are stretched, and the resulting pain and reflex spasm is the basis of Kernig and Brudzinski signs. Spinal rigidity is a more sensitive sign of meningeal irritation than nuchal rigidity, especially in young children. Nuchal stiffness is most readily demonstrated with the child in the sitting position with the legs extended (61). Systemic signs can include pneumonia in children with pneumococcal or H. influenzae meningitis and petechiae, purpura, or signs of DIC in children with meningococcal meningitis. Rash, purpura, and DIC can also be observed occasionally in children with H. influenzae meningitis.
nerve palsies, disturbances in vision, and occasionally papilledema (59). Headache, often accompanied by photophobia, results from inflammation of the meningeal vessels and from increased intracranial pressure. Head retraction, neck stiffness, and spinal rigidity are caused by irritation of the meninges and spinal roots, which elicits protective reflexes intended to shorten the spinal axis and immobilize the irritated tissue (60). With lengthening of the spine, nerve roots are stretched, and the resulting pain and reflex spasm is the basis of Kernig and Brudzinski signs. Spinal rigidity is a more sensitive sign of meningeal irritation than nuchal rigidity, especially in young children. Nuchal stiffness is most readily demonstrated with the child in the sitting position with the legs extended (61). Systemic signs can include pneumonia in children with pneumococcal or H. influenzae meningitis and petechiae, purpura, or signs of DIC in children with meningococcal meningitis. Rash, purpura, and DIC can also be observed occasionally in children with H. influenzae meningitis.
TABLE 7.4 Signs and Symptoms in 39 Infants with Neonatal Meningitis | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Seizures can be generalized or partial in infants or children with bacterial meningitis. Dodge and Swartz reported seizures in 44% of patients with H. influenzae meningitis, 25% of those with S. pneumoniae infections, 10% of those with meningococcal infections, and 78% of those with streptococcal meningitis (62). Focal seizures can be the result of localized involvement of the cerebral hemispheres by bacteria, cytokines, or vascular lesions. In a study by Samson and associates (63), 18% of patients had their first febrile convulsion in the course of meningitis. A lumbar puncture, therefore, is advised in children with their first febrile convulsion, especially in infants or unimmunized children. Lumbar puncture also deserves consideration when febrile convulsions recur and meningitis is suspected (64) or the seizure has atypical features (65). Focal neurologic signs in infants or children with bacterial meningitis can indicate Todds paralysis, ischemic stroke, or sinovenous thrombosis.
Abnormalities of cranial nerves III, IV, and VI can result from local inflammation of the perineurium or impaired vascular supply to the nerves. Transient opsoclonus can occur (66). Cochlear and vestibular deficits occur owing to septic involvement of the endolymphatic and perilymphatic systems of the scala tympani (67) or are caused by cytolytic toxins elaborated by bacteria (68). Conductive hearing loss can result from middle ear dysfunction following meningitis (69). Sensorineural hearing loss, the most common sequela of bacterial meningitis, is more likely in children younger than 1 month or older than 5 years of age, children with hydrocephalus, and those with a decreased CSF glucose (70). Jiang and coworkers suggested that a slightly depressed amplitude of the brainstem auditory potential wave V is a sensitive indicator of brainstem dysfunction in children recovering from meningitis (71). Otoacoustic emissions can detect meningitis-induced sensorineural hearing loss in young infants (72). Selective paralysis of downward gaze can occur (73). In a few rare instances, acute cerebellar ataxia can be the presenting symptom in H. influenzae or N. meningitidis infection (74). Cortical blindness also has been encountered (75).
In young infants, widened sutures can be observed within 2 days after onset of meningitis (76); however, progressive ventricular enlargement can develop without excessive head growth during or after acute bacterial meningitis (77). This enlargement results from cerebral destruction (passive ventriculomegaly) or from the effects of increased intraventricular pressure on already compromised brain parenchyma. In young infants, the low water content of brain parenchyma, lack of myelin, and relatively large subarachnoid spaces permit ventricular enlargement without corresponding increases in head circumference. Cranial bruits over the anterior fontanel and the posterior temporal areas are more common in children with bacterial meningitis than in febrile or healthy controls (72).
Complications
Ventriculitis
Ventriculitis commonly accompanies bacterial meningitis in young infants; in one series, 92% of neonates with fatal meningitis had ventriculitis (79,80). When ventriculitis is accompanied by an obstruction of the aqueduct of Sylvius, the infection can behave like a brain abscess. Rapid increase in intraventricular pressure can induce brainstem herniation and impair perfusion of the periventricular structures. Ventriculitis, treated with high doses of parenteral antibiotics, occasionally requires neurosurgical drainage (80).
Subdural Effusion and Subdural Empyema
Subdural effusions, a complication observed in the majority of young infants with bacterial meningitis, usually occur bilaterally over the frontoparietal region, although localized collections can develop over the occipital region. Effusions are most common when meningitis results from H. influenzae (45% of all effusions); less often, pneumococcus (30% of all effusions) and meningococcus (9% of all effusions) are the responsible pathogens. Subdural effusions result from efflux of intravascular fluids as a consequence of thrombophlebitis of the veins bridging the subdural space, abnormal vascular permeability at the arachnoid–dura interface, or arachnoiditis. Most effusions occur in children younger than 2 years of age. Other factors that favor the evolution of a subdural effusion include a rapid onset of illness, low peripheral white blood count, and high levels of CSF protein and bacterial antigen. Although children with effusions are more likely to have acute neurologic abnormalities or seizures, their outcome is similar to that of children without effusions (81).
Subdural empyema represents infection of the subdural space. In contrast to subdural effusions, subdural empyema rarely complicates bacterial meningitis (82). Indications for suspecting a subdural effusion or subdural
empyema in the presence of meningitis are summarized in Table 7.5 (83). The criteria set forth by Matson more than 30 years ago are still applicable; in particular, the presence of fever that lasts more than 3 to 5 days after the start of antibiotic therapy should prompt initiation of diagnostic studies for these complications. The conditions can be diagnosed best by using gadolinium-enhanced magnetic resonance imaging (MRI).
empyema in the presence of meningitis are summarized in Table 7.5 (83). The criteria set forth by Matson more than 30 years ago are still applicable; in particular, the presence of fever that lasts more than 3 to 5 days after the start of antibiotic therapy should prompt initiation of diagnostic studies for these complications. The conditions can be diagnosed best by using gadolinium-enhanced magnetic resonance imaging (MRI).
TABLE 7.5 Indications for Suspecting a Subdural Effusion in Infants with Purulent Meningitis | ||
|---|---|---|
|
In a subdural effusion, the fluid is xanthochromic or blood-tinged initially and can become less yellow with repeated taps. The protein content ranges between 50 and 1,000 mg/dL and is always higher than the CSF protein obtained simultaneously. Compared with serum, subdural fluid has a disproportionately high albumin-to-globulin ratio. MRI generally shows the fluid to be isointense relative to CSF on both T1- and T2-weighted images. In subdural empyema, the fluid shows a markedly elevated white count and protein concentration, with the glucose concentration being generally less than in CSF (84). On MRI the subdural empyema is hyperintense relative to CSF on proton density-weighted and T2-weighted images. Gadolinium enhancement of the adjacent leptomeninges can be observed in either empyema or effusion.
Subdural effusions can be managed conservatively, and aspiration or surgical intervention is no longer recommended unless empyema is strongly suspected. Most subdural fluid collections resorb spontaneously (81). Surgical intervention can be indicated if the effusion becomes hemorrhagic or causes significant midline shift. The technique for a subdural tap is described by Matson (77). Persistence of subdural effusions can indicate underlying cortical damage (85).
Fluid and Electrolyte Disturbances
Hyponatremia occurs in approximately 20% of patients with meningitis (86). Hyponatremia can be the result of an intracellular shift of sodium with an extracellular shift of potassium as a consequence of the inflammation, the syndrome of inappropriately high secretion of antidiuretic hormone (SIADH), or increased release of atrial natriuretic peptide. Inappropriate ADH release, which can occur with or without clinical hyponatremia, occurs in almost every case of bacterial meningitis and in approximately 60% of cases of viral meningitis (87). Hyponatremia leads to hypervolemia, oliguria, and increased urinary osmolality, despite low serum osmolality. When hyponatremia becomes severe, symptoms can include restlessness, irritability, and convulsions. The diagnosis can be established by measuring serum electrolytes and osmolality and urine output, specific gravity, and osmolality. The condition is treated by restricting free water intake; hypertonic saline solutions can be considered cautiously in children with seizures (88).
Recurrent Meningitis
Recurrent bacterial meningitis can be caused by acquired or congenital anatomic defects, foci of infection, or immunologic disorders. Skull fractures, especially those affecting the base of the brain and extending to the sinuses and petrous pyramids, are the most common cause of recurrent meningitis. They are discussed in Chapter 9. Congenital defects include myelomeningoceles, neurenteric cysts, midline or spinal dermal sinuses, and anomalies of the labyrinthine capsule and stapes foot plate. These conditions are covered in Chapter 5. Neuroschisis that allows entry of bacteria in the CSF can occur as an anterior encephalocele in the nasopharynx or as a small defect in the cribriform or orbital plate. Meningitis caused by these defects can occur in late childhood (89).
Splenectomy, congenital immunodeficiencies such as agammaglobulinemia, or acquired immune disorders such as HIV infection can predispose children or adults to bacterial meningitis. Children with leukemia and lymphoma have a particularly high incidence of recurrent purulent and fungal meningitis (see Chapter 17). Recurrence caused by the same organism can also occur if the initial therapy was inadequate or the organism was resistant to antibiotics (90). The incidence of chronic complications of meningitis is listed in Table 7.6.
TABLE 7.6 Major Complications Seen in 71 Children after Recovery from Meningitis | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Diagnosis
The diagnosis of meningitis requires prompt lumbar puncture and analysis of the CSF (91,92). The technique of lumbar puncture and the interpretation of CSF findings are discussed in the Introduction chapter. Children with focal neurologic deficits, coma, or papilledema require head computed tomography (CT) prior to performing a lumbar puncture (93). When a CT is indicated, a blood culture should be obtained (94) and empiric therapy should be initiated as rapidly as possible while awaiting the imaging study. A brief delay between lumbar puncture and antibiotic treatment may not interfere with isolation of the causative organism.
The gold standard for the diagnosis of bacterial meningitis is identification by culture and Gram stain of CSF. Most bacteria responsible for meningitis can be readily detected by all microbiology laboratories. Culture also provides clinically useful information regarding antibiotic sensitivity. Several bacteria can also be detected by CSF analysis using the polymerase chain reaction (PCR), including the nucleic acids of N. meningitidis (95), H. influenzae, and Mycobacterium tuberculosis (96). PCR can be useful in the diagnosis of meningitis caused by spirochetes (97) and several viruses (98,99) and has considerable promise for detecting bacteria in children with meningitis that is partially treated or due to nonculturable organisms. Partial treatment of bacterial meningitis reduces the sensitivity of culture and Gram stain from 97% to 73% (100) but does not substantially alter the protein or glucose content of the CSF (101).
The CSF in bacterial meningites is usually cloudy and under increased pressure. During the acute stage, polymorphonuclear leukocytes predominate, whereas mononuclear cells appear in the later stages. The cell counts generally range between 1,000 and 10,000 cells/μL, but as many as 6% of cases have few or no cells during the earliest stage of the infection (102). Organisms can be seen intracellularly and extracellularly in smears and can be visualized or cultured in the absence of pleocytosis on rare occasions (103). Eosinophilic leukocytes can be seen in the CSF of patients with parasitic infestations of the CNS, including cysticercosis, trichinosis, toxocariasis, toxoplasmosis, ascariasis, angiostrongyliasis, echinococcosis, gnathostomiasis, or coccidiomycosis (104).
The cerebrospinal fluid typically has a low glucose content in children with bacterial meningitis, often to undetectable levels. In bacterial meningitis, the CSF-to-blood glucose ratio is usually below 0.40 (105). First observed by Lichtheim (106), the decrease was attributed initially to consumption of glucose by bacteria growing in the CSF. Although there remains controversy regarding the cause of hypoglycorrhachia in bacterial meningitis, the finding likely represents a combination of altered transport into the CSF compartment and alterations in glycolysis and the blood–brain barrier (107). Prockop and Fishman observed that the facilitated diffusion of glucose from blood to CSF and from CSF to blood is impaired in bacterial meningitis, although the increase in nonspecific bulk transport of glucose can compensate for the deficit (108). Low CSF glucose also occurs in sarcoidosis, mumps meningitis, herpes zoster meningitis, CNS leukemia, and meningeal carcinomatosis (109).
CSF protein is elevated in 80% to 92% of patients (59), commonly to levels of 100 mg/dL and higher, and some studies show correlations between mortality and increased CSF protein. CSF lactate concentrations can also be increased in bacterial meningitis (110), whereas CSF lactate is normal in aseptic meningitis unless cerebral hypoxia or edema has activated glycolytic pathways (111,112). The slow decrease of CSF lactate to normal with a 3-day course of antibiotic therapy can be useful in the diagnosis of partially treated bacterial meningitis (113) and can aid in differentiating bacterial meningitis from aseptic meningitis (114). The decrease in CSF pH, which also occurs in bacterial meningitis, is more transient than the elevation of CSF lactate and has less diagnostic utility (113).
Neuroimaging studies have important adjunctive roles in identifying the complications of bacterial meningitis, such as hydrocephalus, subdural effusions, or subdural empyema, and in detecting parameningeal abscesses or CSF leaks. Indications for neuroimaging in acute bacterial meningitis include depressed level of consciousness, prolonged, partial, or late seizures, focal neurologic deficits, enlarging head circumference, persistent or recurrent fever during the later stages of treatment, and recurrent meningitis (91,92,93,115). Neuroimaging studies can also improve clinicians’ ability to predict sequelae in children who survive bacterial meningitis. Imaging should be considered strongly in neonates with bacterial meningitis, especially when gram-negative organisms are identified.
In the series of Rennick and colleagues, cerebral herniation occurred in 4.3% of children with meningitis (116). When ventricular dilatation or other imaging signs of increased pressure are present, lumbar puncture should be deferred to diminish the risk of herniation. This is particularly true if the child has a Glasgow Coma Score of 7 or less (117). Severe cerebral edema complicating neonatal meningitis can be associated with uncal herniation (118).
Treatment
Because of the life-threatening nature of bacterial meningitis and the potential for permanent neurodevelopmental sequelae, antibiotic therapy should be instituted as soon as the diagnosis is suspected. The management of the pediatric patients with meningitis is reviewed by Saez-Lorens and McCracken (1), Quagliarello and Scheld (3), Feigin and Pearlman (119), and Wubbel and McCracken (120).
Empiric antibiotic therapy for meningitis in infants younger than 1 month of age consists of therapy with ampicillin, 150 to 300 mg/kg per day in divided doses every 6 to
8 hours, and gentamicin, 2.5 to 7.5 mg/kg per day in one to three divided doses, or cefotaxime, 150 to 300 mg/kg per day divided every 6 to 8 hours, depending on the gestational and postnatal age of the infant (121). Antibiotic therapy should be modified once the identity and sensitivity profile of the pathogen have been determined (Table 7.7). Therapy for group B streptococcal meningitis can consist of ampicillin, as just described, or penicillin G, 250,000 to 450,000 U/kg per day intravenously in three divided doses for infants less than 1 week old and 450,000 U/kg per day in four divided doses for infants older than 1 week. Some add gentamicin, 2.5 to 7.5 mg/kg per day in one to three divided doses. E. coli meningitis can be treated with ampicillin or an expanded-spectrum cephalosporin and gentamicin, as just described. Listeria are not sensitive to cephalosporins, including the expanded-spectrum formulations, so therapy consists of 14 to 21 days of intravenous ampicillin, 150 to 300 mg/kg per day in three to four divided doses, and gentamicin, 2.5 to 7.5 mg/kg per day in one to three divided doses, depending on the infant’s gestational and postnatal age (121). Infants with uncomplicated cases of meningitis require 14 to 21 days of intravenous antibiotic therapy, and complicated cases may require more prolonged treatment.
8 hours, and gentamicin, 2.5 to 7.5 mg/kg per day in one to three divided doses, or cefotaxime, 150 to 300 mg/kg per day divided every 6 to 8 hours, depending on the gestational and postnatal age of the infant (121). Antibiotic therapy should be modified once the identity and sensitivity profile of the pathogen have been determined (Table 7.7). Therapy for group B streptococcal meningitis can consist of ampicillin, as just described, or penicillin G, 250,000 to 450,000 U/kg per day intravenously in three divided doses for infants less than 1 week old and 450,000 U/kg per day in four divided doses for infants older than 1 week. Some add gentamicin, 2.5 to 7.5 mg/kg per day in one to three divided doses. E. coli meningitis can be treated with ampicillin or an expanded-spectrum cephalosporin and gentamicin, as just described. Listeria are not sensitive to cephalosporins, including the expanded-spectrum formulations, so therapy consists of 14 to 21 days of intravenous ampicillin, 150 to 300 mg/kg per day in three to four divided doses, and gentamicin, 2.5 to 7.5 mg/kg per day in one to three divided doses, depending on the infant’s gestational and postnatal age (121). Infants with uncomplicated cases of meningitis require 14 to 21 days of intravenous antibiotic therapy, and complicated cases may require more prolonged treatment.
TABLE 7.7 General Recommendations Regarding Treatment of Meningitis | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
Empiric antibiotic therapy for suspected bacterial meningitis in children older than 1 month of age consists of cefotaxime, 300 mg/kg per day in three or four divided doses, or ceftriaxone, 100 mg/kg per day intravenously divided every 12 hours, and vancomycin, 60 mg/kg per day in four divided doses (121). Vancomycin, used because of potential resistance of S. pneumoniae to penicillin and cephalosporins, should be discontinued as soon as the causative organism is shown to be susceptible to penicillin, cefotaxime, or ceftriaxone. Resistance of S. pneumoniae to penicillin and cephalosporins remains a potential problem in most regions. The prevalence of strains with decreased susceptibility to penicillin approaches 50% in some areas of the United States (122). Penicillin G, 250,000 units/kg per day (maximum dose 12 million units/day), can be used in children or adolescents with meningococcal meningitis.
Children or adolescents with suspected meningitis require droplet and standard precautions for the first 24 hours of appropriate antibiotic therapy. Repeat lumbar puncture should be considered after 24 to 48 hours of therapy to confirm sterilization of the CSF in infants and in children with pneumococcal meningitis who received dexamethasone or have infections with strains that are nonsusceptible to penicillin or cephalosporins. Infants older than 1 month of age, children, and adolescents with bacterial meningitis should receive 7 to 14 days of therapy, depending on the organism and the presence of any complications.
Adjunctive management of infants and children with suspected or proven bacterial meningitis includes control of increased intracranial pressure, treatment of seizures, correction of electrolyte disturbances, treatment of fever, and close monitoring for subdural effusions and severe systemic complications such as DIC, hemorrhagic purpura, or renal failure (123,124). Strategies for the management of childhood bacterial meningitis can be found in the 2003 Red Book of the American Academy of Pediatrics (121).
Dexamethasone therapy, an adjunct in meningitis therapy in infants and children with H. influenzae meningitis and perhaps also in adults with bacterial meningitis, is controversial in children with S. pneumoniae or N. meningitidis meningitis. Clinical trials with dexamethasone in children with H. influenzae meningitis demonstrated a significantly lower incidence of profound hearing loss in the dexamethasone-treated group (125,126,127,128). Recommendations regarding dexamethasone therapy can be found in the most recent edition of the Red Book (121).
Increased intracranial pressure can cause significant alterations in consciousness and can contribute to the morbidity of bacterial meningitis. Intracranial pressure can be reduced by cautious removal of CSF, as might occur with an extraventricular drainage device, or by the use of hyperosmolar agents. These agents can sometimes improve the child’s sensorium promptly. Mannitol is effective for the cytotoxic edema but not for the vasogenic edema of meningitis. Intracranial pressure can be reduced by elevating the head of the patient’s bed by 30 degrees, and pressure spikes associated with suctioning can be minimized by careful sedation. Hyperventilation as a means to lower intracranial pressure by reducing PCO2 may be detrimental (57).
Seizures require prompt treatment using lorazepam, 0.05 to 0.1 mg/kg as needed, followed by loading doses of either phenobarbital, 10 to 20 mg/kg, or fosphenytoin, 15 to 20 mg/kg. Maintenance doses of either phenobarbital or fosphenytoin may be necessary thereafter. Sedation can accompany anticonvulsant therapy. The potential for SIADH in children with bacterial meningitis necessitates close monitoring of fluid therapy, serum electrolytes, and urine output.
Prognosis
The outcome of bacterial meningitis is influenced by the age of the child, the species of the bacterial pathogen, and the duration of disease before the initiation of appropriate antibiotic therapy. In general, the prognosis is less favorable in neonates, regardless of the pathogen, and in older children with pneumococcal meningitis. In some but not all studies (118,129,130), sequelae are more likely in children whose diagnosis and treatment are delayed. Kresky and colleagues observed sequelae in 12% of children whose treatment was begun within 24 hours of the onset of symptoms versus 59% in children who began treatment 3 or more days after the onset of symptoms (131).
In the experience of Thomas, sensorineural hearing loss, the most common sequela of bacterial meningitis, was observed in 8.5% of the surviving children (132); hearing loss was bilateral and severe in 5.6%. Children who survive bacterial meningitis also have learning disabilities, motor problems, speech delay, hyperactivity, blindness, obstructive hydrocephalus, and recurrent seizures. Grimwood and colleagues observed that approximately one in four school-aged meningitis survivors had serious and disabling sequelae, a functionally important behavior disorder, or neuropsychological or auditory dysfunction that adversely affected academic performance (133). Children who have meningitis in the first year of life and survive are at greatest risk for neurodevelopmental sequelae (134).
Hearing loss develops during the first 48 hours of the illness (135) and results from infectious and immune-mediated injury to the cochlea and, occasionally, the semicircular canals. Less often, deafness is caused by an arachnoiditis of the eighth nerve or damage to the auditory projection areas. Recovery of hearing can begin during the first 2 weeks, but hearing can continue to improve for as long as 6 months. However, because hearing loss can be permanent, audiometry is required in all infants, children, and adolescents who recover from bacterial meningitis (136,137).
The 20-year risk for subsequent unprovoked seizures is 13% for patients with early seizures and 2.4% for those without early seizures. When seizures develop, their incidence is highest during the first 5 years after meningitis, but the risk remains elevated during the subsequent 15 years (138). Patients who develop intractable seizures after meningitis incurred before 4 years of age have a high incidence of neocortical seizure foci or mesial temporal sclerosis. The latter group of patients can respond to surgical intervention should seizures remain unresponsive to anticonvulsant therapy (139).
In a meta-analysis of reports of meningitis in children 2 months to 19 years of age published between 1955 and 1993, Baraff and colleagues found that children from developed countries had a lower mortality (4.8% vs. 8.1%) and a lower likelihood of sequelae (17.5% vs. 26.1%) than those from underdeveloped countries (140). Mortality was highest for meningitis caused by S. pneumoniae (15.3%) and lowest for that caused by N. meningitis (7.5%) and H. influenzae (3.8%) (140).
Common Forms of Meningitis
Meningococcal Meningitis
N. meningitidis causes three distinct clinical entities: meningitis, as discussed previously; septicemia, which can precede invasion of the CNS or be an isolated but fulminant form with petechiae and purpura (Waterhouse-Friderichsen syndrome); and chronic meningococcemia, in which an equilibrium between bacteria and host has become established (141). Genetic factors determine, in part, the susceptibility to meningococcal disease. Families with low TNF production have a 10-fold increased risk for fatal outcome, whereas high IL-10 production, a potent inhibitor of TNF (142), increases the risk 20-fold. Children with complement deficiency are also at risk (143), suggesting that screening for complement disorders should be considered in children or adolescents with meningococcal infections.
The meningococcus produces petechial, maculopapular, or morbilliform skin lesions in approximately 75% of patients. Petechiae also are occasionally seen in patients with H. influenzae, pneumococcal, or streptococcal meningitis. Septicemia with N. meningitidis can lead to tissue sensitization and purpura fulminans as a consequence of disseminated intravascular coagulation. Purpura fulminans also has been observed during infections
with other Neisseria species, including N. catarrhalis (144), N. subflava (145), and N. gonorrhoeae (146).
with other Neisseria species, including N. catarrhalis (144), N. subflava (145), and N. gonorrhoeae (146).
Meningococcal septicemia can cause a rapidly evolving fulminant illness with high rates of morbidity and mortality. The pathogenesis of this disorder reflects bacterial embolization and endotoxin-induced shock (147,148,149). Pulmonary microvascular thromboses, composed of platelets and leukocytes, can lead to severe cor pulmonale. Meningococcal endotoxin activates the cascades of procoagulation, anticoagulation, and fibrinolysis as well as the cytokine network and the complement system. Meningococcal endotoxin also produces disseminated intravascular coagulation with rapid consumption of fibrinogen and formation of fibrin thrombi in adrenal glands and renal glomeruli. The fibrin thrombi cause hemorrhagic infarction of the adrenal glands and renal cortical necrosis. Bilateral adrenal hemorrhages occur in two-thirds of fatal cases.
Chronic meningococcemia produces intermittent chills or fever, evanescent rash, joint pain or swelling, or joint effusions. Symptoms can regress without specific therapy or recur over several days or weeks. The rash can assume the form of petechiae, erythema nodosum, papulonecrotic tuberculids, macules, or maculopapules. Recurrent neisserial infections of the nervous system are rare, but they occur with increased frequency in patients with deficiencies of immunoglobulin G (IgG) subclass (150) or complement (151). Complications of meningococcal bacteremia include pericarditis, arthritis, hypopyon, and panophthalmitis (152). Autopsy of 200 fatal cases of meningococcal infection revealed acute interstitial myocarditis with focal necrosis and hemorrhage in 78% (153). Deafness after meningococcal meningitis is more frequent with infections by the uncommon serogroups (W135, X, Y, 29E) than with meningitis caused by serogroup B (154).
Chemoprophylaxis of family members and other intimate contacts is recommended; current information can be found in the Red Book (121,155). Rifampin is the drug of choice in children, whereas rifampin, ceftriaxone, or ciprofloxacine can be used in adults. A quadrivalent vaccine (groups A, C, Y, and W135) is available in the United States for children 2 years of age and older (121). Vaccination of college students who live in dormatories is recommended, given the potential for epidemic meningococcal disease on college campuses (121).
Haemophilus Meningitis
Before the availability of H. influenzae (Hib) vaccine, H. influenzae was the leading cause of meningitis in the United States, causing 8,000 to 11,000 cases annually (1,3,9). Between the years 1985 and 1991 the incidence of H. influenzae meningitis in the United States decreased by 82% (156,157) after the widespread introduction of the vaccine (Table 7.1). H. influenzae vaccines consist of capsular polysaccharides covalently linked to a carrier protein. They are not effective against unencapsulated organisms that are also potential causes of CNS disease (158). Haemophilus meningitis is occasionally caused by types A through F or nontypable species (159,160). Although H. influenzae meningitis occurs almost exclusively in unimmunized children younger than 6 years of age (121), cases have been described in neonates or apparently healthy adults (161).
Hearing loss, the most common neurologic sequela of H. influenzae meningitis, affects approximately 11% of surviving children. Hearing deficits range from mild to profound hearing loss and are more common in children who began treatment 24 hours or more after the onset of symptoms (162). In approximately one-half of the affected children the hearing loss is bilateral. Approximately 15% of survivors have neurologic defects, such as a learning disability, focal neurologic deficits, epilepsy, cortical blindness, or mental retardation (163,164). Mortality rates range from 3% to 5%. Factors associated with poor prognosis include age less than 1 year, illness duration of more than 3 days before therapy, and onset or persistence of seizures after 3 days of treatment (162). Rarely, carotid artery occlusion (165,166), pericarditis, myocarditis, atrioventricular block (167), epiglottitis, cellulitis, and septic arthritis can accompany H. influenzae meningitis (168).
The risk of disease in household contacts is increased considerably during the month after exposure to the index case. Clusters of cases have also been encountered in day care centers. Rifampin prophylaxis has been recommended for index cases during their hospitalization, usually just before discharge, and for adults and children in households of an unvaccinated child younger than 4 years of age, households with an unimmunized child under 1 year of age, and households with an immunocompromised child. Prophylaxis should be given as soon as possible because 54% of secondary cases occur in the first week after hospitalization of the index case (121).
Pneumococcal Meningitis
Predisposing factors for childhood meningitis with S. pneumoniae, the most common cause of meningitis after the neonatal period in populations with compulsory immunization for H. influenzae, include an upper respiratory infection, acute or chronic otitis media, purulent conjunctivitis, and CSF rhinorrhea secondary to developmental abnormalities or trauma (59). Splenectomy, congenital or acquired immunodeficiencies, and disorders that alter splenic function (e.g., sickle cell disease) increase substantially the risk of pneumococcal disease, including meningitis (169,170,171).
The pathogenesis of pneumococcal meningitis differs because the gram-positive organism does not contain endotoxin. Pneumococci adhere to cerebral capillaries using a surface protein, CbpA, that recognizes carbohydrates.
Once attached, invasion involves bacterial recognition of the receptor for platelet-activating factor (PAF) (172). Once in the CSF, a variety of pneumococcal components, including the cell wall, lipoteichoic acid, and several cytotoxins, incite the inflammatory response (173). The cytotoxin pneumolysin contributes to loss of cochlear cells in meningitic hearing loss but does not contribute to cell damage or inflammation in the brain itself (174).
Once attached, invasion involves bacterial recognition of the receptor for platelet-activating factor (PAF) (172). Once in the CSF, a variety of pneumococcal components, including the cell wall, lipoteichoic acid, and several cytotoxins, incite the inflammatory response (173). The cytotoxin pneumolysin contributes to loss of cochlear cells in meningitic hearing loss but does not contribute to cell damage or inflammation in the brain itself (174).
Approximately one-third of children who survive pneumococcal meningitis have sequelae, 19% with hearing loss and 25% with neurologic complications (175). Overall mortality in the series reported by Kornelisse and coworkers was 17%. Coma at admission, respiratory distress, shock, CSF protein level of 250 mg/dL or greater, peripheral white count of less than 5,000 cells/μL, and a serum sodium of less than 135 mEq/L are associated with increased risks of mortality. Of the surviving children described by Arditi and colleagues, 25% had neurologic sequelae, and 32% had unilateral or bilateral hearing loss. In this study the incidence of hearing loss was higher in those who had received dexamethasone (176), illustrating further the controversy associated with dexamethasone therapy in childhood bacterial meningitis.
Staphylococcal Meningitis
Staphylococcus aureus accounts for 0.8% to 8.8% of bacterial meningitis cases in patients of all ages. Approximately 20% of cases occurred in neonates, 10% in children, and 70% in adults (177). As many as 90% of cases occur in association with predisposing factors, such as recent neurosurgical procedures or head trauma. S. aureus causes 12% to 36% of CSF shunt infections, and S. epidermidis causes the majority of the remainder. Meningitis can develop from staphylococcal abscesses within the brain parenchyma or from spinal epidural abscesses. More distant staphylococcal infections, such as oral or abdominal abscesses, sinusitis, osteomyelitis, pneumonia, cellulitis, infected shunts or intravascular grafts, decubitus ulcers, and even abdominal abscesses, have been implicated in occasional cases of S. aureus meningitis. S. aureus is the most likely cause of meningitis in persons with infective endocarditis. Other predisposing factors include immunodeficiency disorders, diabetes mellitus, renal failure, and malignancy (178,179,180). Coagulase-negative staphylococci, especially slime-producing variants (181), can cause indolent infections of ventriculoatrial and ventriculoperitoneal shunts (182,183) (see Chapter 5 for a further discussion of shunt-related infections).
Acute Purulent Meningitis in the Neonate
Neonatal meningitis has an overall incidence of 0.2 to 0.32 cases per 1,000 live births (184). Meningitis occurs more often in male infants (68% to 76%) (185), except for group B streptococcal meningitis, which has a female preponderance (186). Other potential neonatal pathogens include E. coli, Listeria monocytogenes, Citrobacter species, Klebsiella-Enterobacter species, Pasteurella multocida (187), Flavobacterium, Pseudomonas cepacia (188), Salmonella species (189), N. meningitidis (190), and, rarely, H. influenzae. The median age of infants with neonatal meningitis is 10 days (191). Predisposing factors include maternal infections chorioamnionitis, premature rupture of membranes, neonatal urinary anomalies, neural tube defects, and immune dysfunction common among newborns (192,193,194). Infections of the respiratory tract and skin can also predispose to meningitis; bacterial meningitis complicates 20% to 30% of infants with septicemia.
CSF findings can be confusing in neonatal meningitis, given the broad ranges of normal values for cell count and protein content (194,195,196,197,198,199). White blood counts of less than 32 cells/μL occur in 29% of group B streptococcal and 4% of gram-negative neonatal meningitis; protein concentrations of less than 170 mg/dL occur in 47% and 23%, and CSF-to-blood glucose ratios of grerater than 0.44 occur in 45% and 15%, respectively (198). CSF values for healthy term and preterm infants are presented in the Introduction chapter.
Since the late 1970s group B Streptococcus (S. agalactiae) has been an extremely important pathogen in the neonate (200,201,202). The two clinically and epidemiologically distinct forms of group B streptococcal disease are now recognized as a paradigm for several bacterial pathogens that infect the neonate. In the first, termed “early-onset disease,” symptoms begin within 10 days of birth. The infants are often seriously ill with respiratory distress, shock, or DIC at the time of diagnosis, and the mortality is quite high (58%). In this form of infection, bacteria can be isolated often from CSF, blood, and other fluids or tissues. In the second clinical pattern, termed “late-onset disease,” symptoms begin after 10 days of age. The infants are less severely ill, and death or permanent neurologic sequelae are less frequent (19%). Organisms are usually isolated from the CSF or blood only. However, infants with late-onset disease with several bacteria, including group B Streptococcus, can have osteomyelitis, which can cause pseudoparalysis of the upper or lower extremities.
The diagnosis of group B neonatal meningitis can be confirmed by lumbar puncture. Blood cultures are positive in 50% to 90% of infants with early-onset meningitis (203,204), and cultures of the nasopharynx may yield the causative organism in as many as 40% of cases. Antigen detection systems for analysis of CSF or urine have variable sensitivity and specificity and are generally not recommended. The prognosis of neonatal meningitis remains discouraging despite 21 to 28 days of therapy with the available antimicrobials (Table 7.7). In the series
of Hristeva and coworkers the mortality was 26% with neurologic sequelae in 27% of survivors (204). Most experts recommend repeat CSF analysis after 48 hours of therapy to ensure eradication of bacteria from the CSF of infants with neonatal meningitis (121).
of Hristeva and coworkers the mortality was 26% with neurologic sequelae in 27% of survivors (204). Most experts recommend repeat CSF analysis after 48 hours of therapy to ensure eradication of bacteria from the CSF of infants with neonatal meningitis (121).
Chronic and Granulomatous Bacterial Meningitis
Tuberculous Meningitis
Despite advances in chemotherapy, meningitis caused by Mycobacterium tuberculosis remains a serious worldwide pediatric problem, and even in the United States, tuberculous meningitis causes more deaths than any other form of tuberculosis. In England and Wales, tuberculous meningitis occurred in 4.4% of children reported during 1983 to have tuberculosis (205).
Pathology and Pathophysiology.
In children, tuberculous meningitis almost always accompanies generalized miliary tuberculosis. In an older clinical series published in 1997 (206), 68% of children with meningitis had miliary tuberculosis, and conversely, 81% of children with miliary disease developed tuberculous meningitis. In a Turkish series published in 1998, miliary tuberculosis was seen by chest radiography in 20% (207).
In the majority of cases in the United States, infection is caused by the human type of mycobacteria. Bovine tuberculosis has become uncommon because of the widespread pasteurization of milk. The human form also is more common in tropical countries because of the relative inaccessibility of nonhuman milk and milk products.
Involvement of the meninges is probably secondary to a small tuberculoma in the cortex or the leptomeninges (1), and the brains of most patients who die of tuberculous meningitis demonstrate older superficial foci. Tuberculomas of the choroid plexus are a less common site for the infection.
At autopsy, the meninges look gray and opaque, and a gelatinous exudate fills the basal cisterns, particularly the anterior portion of the pons. Small tubercles can appear over the convexity of the brain or the periventricular area. The basal ganglia and thalamus in the region of the lenticulostriate and thalamo-perforating arteries are involved in 46% of cases (208).
Microscopic examination shows the exudate to consist of lymphocytes, plasma cells, and large histiocytes with typical caseation. Langhans giant cells are rare. The meningeal arteries are inflamed and thrombosed, and secondary infarctions of the superficial cortex are common. Tuberculomas are now relatively rare in Western countries, but were at one time one of the most common causes for a posterior fossa mass in children (209). They occur most commonly in the cerebellum and less often within the brainstem.
Clinical Manifestations.
The most common form of tuberculous meningitis, constituting approximately 70% of all cases, is a caseous meningitis that results from direct invasion of the meninges. In approximately 75% of children, this invasion occurs while the primary focus is fresh (i.e., less than 6 months old). The illness is often precipitated by an acute infectious disease, commonly measles. Before 1955, its incidence in the United States was highest in the spring. When tuberculous meningitis is part of the initial attack, its incidence is highest among children between the ages of 1 and 2 years. When it is a complication of systemic tuberculosis, it is more likely to affect older children.
Untreated, tuberculous meningitis rapidly progresses to death, with an average duration of only 3 weeks. Lincoln distinguished three stages of the illness, each lasting approximately 1 week (210).
In the initial stage (stage 1), gastrointestinal symptoms predominate, and no definite neurologic manifestations are seen. The child can be apathetic or irritable with intermittent headache, but the results of the neurologic examination are negative. In approximately 10% of patients, commonly in infants, a febrile convulsion is the most significant symptom of this stage.
During stage 2, the child develops drowsiness and disorientation, with signs of meningeal irritation. The deep tendon reflexes are hyperactive, abdominal reflexes disappear, and ankle and patellar clonus can be present. Cranial nerve signs are evident, with involvement of cranial nerves VII, VI, and III, in that order of frequency (207). Choroid tubercles are present in 10% of patients.
During stage 3, the patient is comatose, although periods of intermittent wakefulness can occur. The pupils are fixed, and recurrent clonic spasm of the extremities, irregular respiration, and a rising fever are present. Hydrocephalus develops in approximately two-thirds of patients whose illness lasts longer than 3 weeks. It was apparent at presentation in 80% of patients in the Turkish series of Yaramis and coworkers (207). It is particularly common when treatment is delayed or inadequate.
A serous form of tuberculous meningitis is encountered less commonly. In children known to have active primary tuberculosis, the presenting symptom is meningeal irritation. Unlike in tuberculous meningitis, the CSF is normal.
A third form of tuberculous meningitis, seen in 17% of instances, consists of cases with primary spinal cord infection. These children can have major problems with blockage of the spinal canal that can be associated with Pott disease. Spinal tuberculosis can be differentiated from the other types by its history and laboratory findings. The symptoms usually last longer, often existing for 6 months before meningitis is considered. A fall often produces back pain and staggering or clumsy gait. Abdominal pain, presumably of root origin, is common. Nuchal or spinal rigidity can develop while the sensorium remains clear. CSF can be scant and show marked elevation in protein.
Recognition of tuberculous meningitis can be difficult because the characteristic signs and symptoms of meningitis can be absent during the early stages of the illness. In the series of Idriss and coauthors, nuchal rigidity occurred in 77%, apathy in 72%, fever in 47%, vomiting in 30%, drowsiness in 23%, headache in 21%, coma in 14%, convulsions, sweating, facial palsy, optic atrophy, and abducens and oculomotor palsies each in 9%, hemiparesis in 5%, and eighth-nerve palsy and diabetes insipidus each in 2% (211).
Metabolic disturbances during tuberculous meningitis include metabolic alkalosis, hyponatremia, hypochloremia, and hypotonic expansion of extracellular fluid. Intracellular potassium concentration is normal or decreased, whereas sodium concentration within red cells and skeletal muscle generally increases. No evidence exists for a salt-losing renal lesion in tuberculous meningitis, although hypervolemia can result in secondary hypoaldosteronism and increased secretion of atrial natriuretic peptide factor (212). The hyponatremic syndrome in tuberculous meningitis is associated with elevations in atrial naturietic peptide more often (65%) than elevation of vasopressin (3%) (212). Vomiting often tends to aggravate electrolyte disturbances.
The presence of focal neurologic signs in a patient with tuberculous meningitis and with hypoglycorrhachia persisting beyond 12 weeks should suggest a tuberculous brain abscess or tuberculoma (213,214). Tuberculomas or vascular occlusions can develop weeks or months after active therapy is instituted (215). However, they were seen in only 2% of patients in the series of Yaramis and colleagues (207). The presence of these complications indicates the need for revision of chemotherapy. Paradoxic expansion of intracranial tuberculomas can occur during therapy (216).
Diagnosis.
The diagnosis of tuberculous meningitis depends on the clinical picture of a subacute meningitis, a history of exposure to the disease, often from an otherwise asymptomatic older relative, a positive skin test result, and the CSF changes.
Anergy to tuberculosis can occur in up to 36% of patients (218). In the experience at Bellevue Hospital, New York City, 85% of patients had positive reactions. Another 10% were inadequately tested or died before tuberculin tests could be completed (210,219). In the Turkish series, only 30% had a positive skin test result (207). In the series of Kent and coworkers, which included both children and adults, 75% of patients reacted positively to 100 U or less of purified protein derivative (220). A decreased humoral immune response can occur in the presence of intact cell-mediated immunity (221). The chest radiograph is not always reliable; in the series of Zarabi and colleagues, children with tuberculous meningitis had normal chest radiograph results in 43% of cases, signs of disseminated miliary tuberculosis in 23%, and calcifying primary intrathoracic tuberculosis in 10% (222). In the more recent series of Yaramis and colleagues, the chest radiograph result was abnormal in 87%; a hilar adenopathy was the most common abnormality, being present in 34% (207).
The CSF findings are characteristic. The fluid has a ground glass appearance and when spun down forms a pellicle in which the organisms can occasionally be visualized. In 85% of cases the total CSF cell count was less than 500 cells/μL (207). The cells are composed of reactive mononuclear and ependymal cells with few polymorphonuclear cells. CSF protein is invariably increased (207,220). Of tuberculous meningitis patients reported from Turkey, 83% showed a sugar of 40 mg/dL or less (207). In the Australian series of Kent and colleagues, the initial CSF glucose was normal in 60%, but in the majority of patients (74%) who initially had a normal CSF glucose level, subsequent values fell below normal (220). Tuberculous meningitis can be diagnosed rapidly by means of the polymerase chain reaction (PCR), enzyme-linked immunosorbent assay (ELISA), and latex particle agglutination detection of mycobacterial antigen (159,223,224). ELISA detection of IgG and IgM antibodies and identification of tuberculostearic acid in CSF with gas chromatography and mass spectrometry also can assist in arriving at a diagnosis of tuberculous meningitis (225). CSF culture results for mycobacteria are positive in less than one-half of instances (207). Performing repeated lumbar punctures and obtaining large volumes of CSF markedly enhance the likelihood of obtaining a positive culture result (226). The PCR for early detection of M. tuberculosis in CSF has a sensitivity of only 48%; microscopy has a sensitivity of 9% (159). Fatal Mycobacterium avium meningitis has been confused with meningitis caused by M. tuberculosis in immunocompetent children (227).
Diffuse or focal electroencephalographic (EEG) abnormalities occur in approximately 80% of cases (228). Neuroimaging is of relatively little assistance in the diagnosis of tuberculous meningitis. Cranial CT within 1 week of initial symptoms can reveal basilar enhancement, ventricular dilatation, or infarction (207,229). MRI with gadolinium enhancement also can demonstrate meningeal inflammation. Mild to marked third ventricular enlargement is extremely common and was invariably present in one study (230). Severe hydrocephalus was demonstrable by CT scans in 87% of children with tuberculous meningitis but in only 12% of adults. Visible infarcts are present in 28% to 40% of cases, usually involving the territory of the middle cerebral artery. Patients with nonenhancing lesions have a good prognosis, whereas those with enhancing lesions are likely to die or have irreversible sequelae despite medical therapy or shunting (205,230).
The appearance of tuberculomas revealed by neuroimaging studies differs from that of abscesses, metastases, and gliomas. MRI of tuberculomas shows large, ring-enhancing lesions with low intensity on T2-weighted images and intermediate intensity on T1-weighted images. Small lesions with ring enhancement on CT scan show a central bright signal on T2-weighted images with a peripheral low-intensity rim, surrounded by high-intensity edema (231).
Treatment.
Therapy for tuberculous meningitis must be prompt and adequate (232). It includes appropriate chemotherapy, correction of fluid and electrolyte disturbances, and relief of increased intracranial pressure.
Treatment of tuberculous meningitis, as recommended by the Committee on Infectious Diseases of the American Academy of Pediatrics (233), should include daily doses of four drugs for 2 months: isoniazid (INH), rifampin, pyrazinamide, and streptomycin. This is followed by INH and rifampin administered daily or twice weekly under direct observation for 10 months. For patients who may have acquired tuberculosis in geographic locales where resistance to streptomycin is common, capreomycin (15 to 30 mg/kg per day) or kanamycin (15 to 30 mg/kg per day) may be used instead of streptomycin. Treatment of tuberculomas should be similar to that for tuberculous meningitis. CSF penetration of INH (89%) and pyrazinamide (91%) changes little during the course of therapy for tuberculous meningitis; penetration of rifampin (5%) and streptomycin (20%) can decline to result in subtherapeutic levels. Corticosteroids have little effect on the penetration of these drugs (234).
INH (10 to 15 mg/kg per day up to a maximum dose of 300 mg/day, 20 to 40 mg/kg per dose twice weekly up to a maximum of 900 mg per dose) is a bactericidal and bacteriostatic drug that interacts with nucleic acid synthesis and inhibits production of cell wall mycolic acid (235). Peak serum concentrations usually range from 6 to 20 μg/mL. The optimal duration of treatment with INH is not known, but therapy should be continued for at least 1 year. Peripheral neuritis can complicate the use of INH in some patients. This complication can be prevented or relieved by pyridoxine (25 to 50 mg/day). Adding pyridoxine is probably not necessary for infants or children unless they are malnourished, but it is recommended for adolescents (232). Other adverse reactions can include gastrointestinal and hematologic hypersensitivity and lupus-like reactions. Hepatitis is rare in children younger than 11 years of age. When INH is used together with rifampin, the incidence of hepatotoxicity increases if the INH dose exceeds 10 mg/kg per day.
Rifampin (10 to 20 mg/kg up to 600 mg daily or twice weekly) is a bactericidal drug that inhibits RNA synthesis by binding to bacterial DNA-dependent RNA polymerase. Rifampin is recommended for children with resistant organisms. Adverse reactions include gastrointestinal and hematologic hypersensitivity, most often thrombocytopenia, flulike symptoms, and hepatitis. Birth control pills may be rendered ineffective.
Pyrazinamide (20 to 40 mg/kg per day or 50 to 70 mg/kg twice weekly up to 2,000 mg/day) is a bacteriostatic drug used for drug-resistant organisms. It can cause hepatotoxicity, hyperuricemia, arthritis, skin rash, and gastrointestinal upset.
Streptomycin (20 mg/kg intramuscularly daily or twice weekly up to 1,000 mg/day) is a bactericidal drug that blocks protein synthesis. Adverse reactions include ototoxicity with greater incidence of vestibular than auditory involvement, hypersensitivity, and, rarely, nephrotoxicity. Streptomycin toxicity usually develops after 12 weeks of therapy.
Ethambutol (15 to 25 mg/kg per day or 50 mg/kg per dose twice weekly up to 2,500 mg/day) is a bacteriostatic drug that has replaced para-aminosalicylic acid in older individuals. It is usually reserved for children older than 5 years of age because of the risk of optic neuritis or optic atrophy in younger children. These complications are rare if the recommended dose is not exceeded. Impairment of visual acuity or color vision may occur, as well as rashes, gastrointestinal symptoms, and fever.
Para-aminosalicylic acid (200 mg/kg per day in three divided doses up to 12 g/day) reduces the emergence of drug-resistant strains. Gastrointestinal side effects can be intolerable.
In the United States, the incidence of drug-resistant strains has been low, but is becoming an increasing problem, and in some regions of the world resistance to INH can be as high as 30%. This percentage makes a laboratory evaluation of drug susceptibility imperative. Ethionamide, cycloserine, kanamycin, amikacin, ofloxacin, ciprofloxacin, and capreomycin should be considered in multidrug-resistant tuberculosis (236).
Spinal arachnoiditis is a common complication of tuberculous meningitis. Intrathecal hyaluronidase has been reported in a nonrandomized trial to reduce the mortality and disability of patients with tuberculous meningitis when compared with a control group (217).
Adjuvant treatment with corticosteroids such as prednisone remains controversial. In the study of Schoeman and coworkers (237), prednisone improved the survival rate and intellectual outcome of patients with tuberculous meningitis; it did not affect the incidence of motor deficits, blindness, deafness, increased intracranial pressure, or basal ganglia infarction (237). Its use has been recommended for seriously ill patients (233,238). In other studies, corticosteroids reduced intracranial pressure, basal exudate, and cerebral edema without altering morbidity or mortality (234,239) and relieved pulmonary symptoms in miliary tuberculosis (232). The neurologic state of patients sometimes deteriorates shortly after the initiation
of therapy. Corticosteroid treatment can benefit this condition and can favorably influence mortality if started before the onset of coma (238,240). Most experts consider 1 to 2 mg/kg per day of prednisone or its equivalent given for 6 to 8 weeks to be appropriate (233).
of therapy. Corticosteroid treatment can benefit this condition and can favorably influence mortality if started before the onset of coma (238,240). Most experts consider 1 to 2 mg/kg per day of prednisone or its equivalent given for 6 to 8 weeks to be appropriate (233).
Intracranial pressure above 400 mm of CSF significantly impairs perfusion of cerebral tissue, and patients who have consistent severe elevations of intracranial pressure require short courses of dexamethasone (6 mg/m2 every 4 to 6 hours) to reduce edema or neurosurgical intervention with ventriculo-external or ventriculoperitoneal shunts to relieve acute hydrocephalus (241,242). Hydrocephalus becomes more marked at pressures up to 600 mm CSF, by which point hemoperfusion has fallen to approximately 40% of normal. Peacock and Deenny observed that ventriculoperitoneal shunting of patients with tuberculous meningitis resulted in a better outcome than was seen in matched nonshunted children (243).
The treatment of tuberculoma has been considered by Bhagwati and Parulekar. In their experience, some 60% of the patients responded to drugs alone and 23% required surgical intervention for failure to respond or mass effect (244). Infratentorial tuberculomas associated with hydrocephalus should receive ventriculoperitoneal shunts.
Prognosis.
Untreated tuberculous meningitis was invariably fatal. With the newer treatment regimens, fewer survivors have major sequelae than were observed several decades ago (245). Even so, in the experience of Deeny and coworkers, reported in 1985, only 18% of survivors were neurologically and intellectually normal (246). As a rule, the prognosis is good for patients presenting with stage 1 disease and generally poor for those presenting with stage 3 disease. In the series of Kent and colleagues 25% of patients presenting with stage 2 disease died or were left with sequelae (220). With treatment, CSF glucose returns to normal within 1 to 2 months and CSF protein becomes normal within 4 to 26 months (220).
Major neurologic sequelae include hydrocephalus, spastic pareses, seizures, paraplegia, and sensory disturbances of the extremities. Late ophthalmologic complications include optic atrophy and blindness. Hearing and vestibular residua can occur as a consequence of both streptomycin therapy and the disease process itself. Minor neurologic sequelae include cranial nerve palsies, nystagmus, ataxia, mild disturbances of coordination, and spasticity.
Intellectual defects occur in approximately two-thirds of survivors (246). These patients have a high incidence of encephalographic abnormalities, which correlate with the persistence of sequelae such as convulsions and mental subnormality. Intracranial calcifications develop in approximately one-third of children who recover from tuberculous meningitis (247). Panhypopituitarism has been reported after cured tuberculous meningitis (248). One-fifth of survivors have such selective pituitary hypothalamic disturbances as sexual precocity, hyperprolactemia, and deficiencies in ADH, growth hormone, corticotropin, and gonadotropin (249).
BRAIN ABSCESSES
Brain abscess is a relatively uncommon but life-threatening infection in children (264). A brain abscess consists of localized free or encapsulated pus within the brain substance. Although the condition has been known for more than 200 years and its association with cyanotic congenital heart disease was described more than 100 years ago, the absence of classic signs and symptoms hinders its early clinical diagnosis and treatment.
Etiology and Pathogenesis
Predisposing contiguous site or distant pathologic states are always present. In the experience of Ersahin and coworkers, reported in 1994, the cause of the abscess was meningitis in 36% of cases, otitis in 27%, head injury in 16%, and congenital heart disease in 9%. Multiple abscesses occurred in 29% of the children (265). However, congenital heart disease was the most common predisposing factor between 1981 and 2000 and from 1945 to1980 at Children’s Hospital Boston (266). Pyogenic organisms gain access to the brain substance by one of three routes. The first is through the bloodstream either from a remote infection, as a consequence of sepsis, or in association with a cardiopulmonary malfunction, most commonly cyanotic congenital heart disease with a right-to-left shunt. The second is by extension of contiguous infections such as leptomeningitis, infections of the middle ear, or infections of the paranasal sinuses either directly or as a result of septic thrombophlebitis of bridging veins. The third is as a complication of a penetrating wound. Children with chronic bacterial infection are more prone to this complication, in that they can develop an infected infarct (267).
The most common causative organisms in order of frequency are anaerobic and microaerophilic streptococci, Fusobacterium species, β-hemolytic streptococci,
S. aureus, and pneumococci. Actinomyces (gram positive) as well as Bacteroides and other gram-negative rods, particularly Haemophilus species, are seen less often (268,269). Abscesses caused by Aspergillus and Nocardia (270) or protozoa such as Toxoplasma (271) are even rarer in the nonimmunocompromised host. Anaerobic organisms alone cause approximately 56% of brain abscesses, aerobic bacteria alone cause approximately 18%, and mixed aerobic and anaerobic bacteria cause approximately 26% (272,273). Frequently, the organism cannot be identified, often owing to failure to perform the appropriate cultures or the use of antibiotics before securing cultures.
S. aureus, and pneumococci. Actinomyces (gram positive) as well as Bacteroides and other gram-negative rods, particularly Haemophilus species, are seen less often (268,269). Abscesses caused by Aspergillus and Nocardia (270) or protozoa such as Toxoplasma (271) are even rarer in the nonimmunocompromised host. Anaerobic organisms alone cause approximately 56% of brain abscesses, aerobic bacteria alone cause approximately 18%, and mixed aerobic and anaerobic bacteria cause approximately 26% (272,273). Frequently, the organism cannot be identified, often owing to failure to perform the appropriate cultures or the use of antibiotics before securing cultures.
TABLE 7.8 Some Rarer Organisms Responsible for Meningitis | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Pathology
The earliest stage of an abscess is cerebritis (septic encephalitis). This is usually localized in the white matter even when infection has to pass through the cortex. It is an edematous area with softening and congestion of brain tissue, often with numerous petechial hemorrhages. The center becomes liquefied, thus forming the abscess cavity (Fig. 7.3). Initially, its wall is poorly defined and irregular. Gradually, a firmer, thicker wall develops, which ultimately can become a thick, firm, concentric capsule of fibrous tissue. The adjacent brain tissue is infiltrated with polymorphonuclear leukocytes and plasma cells near the abscess, with lymphocytes in the more peripheral zones, and heavy lymphocytic cuffing of vessels in the area. In more chronic abscesses, a border of granulation tissue merges gradually into a collagenous capsule. Abscesses enlarge within the softer and less vascular white matter and extend toward the ventricles, rupturing into this space, rather than into the subarachnoid fluid. Seeding of the leptomeninges and subsequent meningitis results from local thrombophlebitis more often than from actual rupture.
Abscesses resulting from hematogenous spread can be localized within any part of the brain, but most commonly occur in the distribution of the middle cerebral artery at the junction of gray and white matter of the cerebral
hemispheres. By contrast, abscesses derived from contiguous sources tend to be superficial and close to the infected bone or dura. Multiple abscesses occur in 6% of patients, usually when sepsis or congenital heart disease is the predisposing cause.
hemispheres. By contrast, abscesses derived from contiguous sources tend to be superficial and close to the infected bone or dura. Multiple abscesses occur in 6% of patients, usually when sepsis or congenital heart disease is the predisposing cause.
 FIGURE 7.3. Anterior frontal lobe abscess in a child with cyanotic congenital heart disease. The brain is edematous and flattening of the gyri is seen. Gross representation of coronal section. (Courtesy of Dr. Hideo H. Itabashi, Departments of Pathology and Neurology, Los Angeles County Harbor Medical Center, Torrance, CA.) |
Clinical Manifestations
During initial stages of the brain abscess, the clinical picture can be nonspecific. The patient, who can have cyanotic congenital heart disease or a primary infection, develops the complex of headache, vomiting, and convulsions, either partially or in its entirety. As the abscess progresses, the neurologic signs, which initially were minimal or completely absent, become readily apparent. As indicated in Table 7.9 (274), they include papilledema, lateralizing signs, notably hemiparesis or homonymous hemianopia, and more obvious indications of increased intracranial pressure. Focal percussion tenderness often aids in the localization of the lesion. Untreated, the condition is usually fatal, either as a consequence of decompensated increased intracranial pressure or from sudden rupture of the abscess into the ventricular system. Sudden rupture is marked by a sudden high fever, meningeal signs, and deterioration of consciousness (275).
Cerebellar abscesses are most commonly seen in association with mastoid infections and occur most frequently during the second and third decades of life. Symptoms and neurologic signs are those associated with any posterior fossa mass lesion. Mastoid infections also give rise to abscesses of the temporal lobe. Abscesses of the pituitary fossa can have a prolonged course that simulates a slow-growing tumor (276).
TABLE 7.9 Incidence of Symptoms and Neurologic Findings in 19 Cases of Brain Abscess with Congenital Heart Disease | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Intramedullary abscesses of the spinal cord have been observed in infants as young as 8 days of age. In a review of 73 cases, 17% were found to have occurred before the age of 5 years (277).
Diagnosis
Peripheral blood counts are usually of little value in establishing the diagnosis of brain abscess. Peripheral white blood cell counts of greater than 11,000 cells/μL occur in only one-third of patients (278). The erythrocyte sedimentation rate is normal in 11% of patients. The CSF leukocyte count in patients without congenital heart disease is greater than 5 cells/μL in 80% of cases. In patients with congenital heart disease, the CSF cell count is normal in 47% and the CSF protein is normal in 67% (278). Generally, the more striking the CSF pleocytosis, the closer the abscess is to the ventricular lining. Hypoglycorrhachia of less than 45 mg/dL occurs in approximately 30% of cases. Bacterial culture results of CSF are usually negative unless the abscess is leaking into the ventricular system (278). As a rule, a lumbar puncture should be avoided in children with brain abscess because the procedure results in neurologic deterioration in 15% to 33% of cases and the information is of little use in management (278).
 FIGURE 7.4. Brain abscess. A: Axial T2-weighted image showing a hyperintense lesion with hypointense rim and surrounding vasogenic edema in the posterior left frontal lobe. B: Sagittal postcontrast T1-weighted image demonstrating hypointense central portion and rim enhancement of the mass. C: The lesion is very bright on the axial diffusion-weighted image. D: Corresponding apparent diffusion coefficient map revealing the low signal intensity of the mass, consistent with restricted diffusion of water molecules. Surrounding vasogenic edema is hyperintense due to increased diffusion. |
The EEG almost always shows a focal slowing, which corresponds closely to the location of the abscess. Neuroimaging studies (CT and MRI) are essential diagnostic tools for making an accurate diagnosis (279) (Fig. 7.4). These reveal a focal process that rapidly assumes the character of a mass lesion with a contrast-enhancing margin (279). If MRI is not available, CT or cerebral arteriography is the preferred procedure when the abscess requires further delineation (280). Focal arterial displacement and the presence of the ripple sign, a concentric, curvilinear
displacement of opacified sulci in the late arterial phase caused by perifocal edema, are helpful arteriographic findings (281). Hyperintensity on diffusion-weighted imaging (DWI) with a reduced apparent diffusion coefficient (ADC) is characteristic of brain abscess (Fig. 7.4); in the absence of restricted diffusion on DWI, in vivo proton magnetic resonance spectroscopy can distinguish brain abscesses from cystic tumors (282,283,284,285).
displacement of opacified sulci in the late arterial phase caused by perifocal edema, are helpful arteriographic findings (281). Hyperintensity on diffusion-weighted imaging (DWI) with a reduced apparent diffusion coefficient (ADC) is characteristic of brain abscess (Fig. 7.4); in the absence of restricted diffusion on DWI, in vivo proton magnetic resonance spectroscopy can distinguish brain abscesses from cystic tumors (282,283,284,285).
In patients with cyanotic congenital heart disease, a brain abscess must be differentiated from intracranial vascular accidents and hypoxic attacks. Thromboses of arteries, veins, and dural sinuses are particularly common in severely cyanotic infants. Although dehydration and stasis play a minor role in the evolution of cerebral vascular thromboses, in the series of Tyler and Clark virtually every infant who experienced a thrombosis had an arterial oxygen saturation of 10 volumes percent or less (286). Except for a more abrupt onset of symptoms, the clinical picture can mimic that of an abscess; however, the patient’s age is of considerable importance in the differential diagnosis. Brain abscesses are rare before 2 years of age, whereas vascular thromboses are rare in patients older than 2 years (286). Emboli in patients with congenital heart disease are highly unusual, and when present they are precipitated by cardiac or other surgery. The differential diagnosis of these two entities and their distinction from hypoxic attacks, which are often accompanied by headaches, is further covered in the section on neurologic complications of cardiac disease in Chapter 17.
A partially treated meningitis also should be considered in the differential diagnosis of a brain abscess. It lacks the neurologic and EEG evidence of focal central involvement, and, of course, neuroimaging studies are diagnostic.
Clark pointed out that old focal neurologic signs, which are often present in the patient with cyanotic congenital heart disease, are aggravated by stress, including a recent infection (287). The ensuing picture simulates a progressive lesion.
Treatment
Treatment of any brain abscess should include a diligent search for the source of infection (288,289). In a child without congenital heart disease, the presence of multiple brain abscesses should raise the question of immunodeficiency. Fungal brain abscesses in immunocompromised patients have death rates in excess of 95% (290). Once the diagnosis has been made, proper selection of antimicrobial agents with good penetration of the CNS should follow. Initial therapy should include coverage for anaerobic as well as aerobic organisms. The antibiotics of choice are generally intravenous penicillin G, chloramphenicol, and metronidazole. Oxacillin or methicillin also should be started until it is established that a β-lactamase–producing organism is not involved. Ampicillin can be used, but is not as effective as penicillin against anaerobic organisms. Surgical drainage should be performed without delay, and antibiotic therapy should be withheld until immediately after cultures have been obtained (278,291,292,293). In the early phase of cerebritis, however, infection can respond to antibiotic therapy alone. Subsequent therapy is guided by laboratory determination of antibiotic sensitivity and by neuroimaging studies (294). It is continued for a minimum of 3 weeks and often for 4 to 6 weeks. Intravenous fluid is usually limited to 1,500 mL/m2 to minimize cerebral edema.
Controversy surrounds the indications for surgical therapy and the various surgical methods. These methods include tapping of the abscess, with or without excision of the shrunken abscess capsule, and operative removal of the acute abscess (295). Generally, surgical therapy is indicated when the patient does not improve after 24 hours of antibiotic therapy, when clinical status deteriorates, or when there is life-threatening displacement of cerebral structures. Sinusitis or mastoiditis contiguous to a brain abscess should be treated vigorously, usually with prompt surgery. A short course of corticosteroids to reduce life-threatening edema can be used safely.
When an abscess is fully established, surgical treatment is usually necessary. Mampalam and Rosenblum compared treatment by surgical excision with treatment by aspiration (278). Excision resulted in a shorter course of antibiotic therapy, but there was no difference in morbidity or mortality. Wright and Ballantine recommended total excision of brain abscesses, having had no deaths with this approach (296), whereas significant mortality was associated with drainage or medical treatment alone. Berlit and coworkers recommended burr hole aspiration plus antibiotic treatment of brain abscesses. In their study, mortality for this group was 9%, as opposed to 62% for the subgroup treated with antibiotics alone (297). The affected region is aspirated and the aspirate examined for aerobic and anaerobic bacteria, mycobacteria, fungi, and parasites, notably Toxoplasma. Patients with increased intracranial pressure should not receive halothane, trichloroethylene, or methoxyflurane because these agents can cause considerable increase in intracranial pressure (298). Because seizures can occur owing to the cortical injury produced by the brain abscess or its surgical therapy, anticonvulsant medication should be used during the acute phase.
Complications
Residual defects include seizure activity, localized neurologic abnormalities, mental retardation, and hydrocephalus. Intracranial complications of frontal sinusitis (Pott’s puffy tumor) include hemiparesis, obtundation, and
aphasia (301). Seizures can appear at any time between 1 month and 15 years after a supratentorial abscess, with the latent period being somewhat shorter when the abscess occurs in the temporal lobe. In 40% of patients, seizures develop within 1 year, and the mean time of onset is 3.3 years. No relationship exists between the development of seizures and the age of the patient or the mode of surgical therapy (302).
aphasia (301). Seizures can appear at any time between 1 month and 15 years after a supratentorial abscess, with the latent period being somewhat shorter when the abscess occurs in the temporal lobe. In 40% of patients, seizures develop within 1 year, and the mean time of onset is 3.3 years. No relationship exists between the development of seizures and the age of the patient or the mode of surgical therapy (302).
The overall mortality has declined from 30% in the pre-CT era to almost none in the past few years. The prognosis remains poor in infants with brain abscesses, with mortality approaching 50% (303).
Subdural Abscesses
Subdural abscesses or subdural empyema result from sepsis, from a direct extension of an infection such as an osteomyelitis, or from bacterial contamination of a subdural effusion in meningitis or a post-traumatic subdural hematoma. Approximately two-thirds occur subsequent to a prior craniotomy. Otitis media and pneumococcal meningitis are other sources of organisms for subdural abscesses in children, whereas sinusitis is a more common antecedent in adults. Approximately 9% of children with brain abscesses develop subdural empyemas. These lesions generally occur over the convexity of the brain, but 12% are found interhemispherically.
Signs and symptoms are those of a space-occupying lesion in association with evidence of infection. Increased intracranial pressure and focal neurologic findings, notably focal seizures, are common (304,305).
The presence of subdural abscesses is demonstrated by neuroimaging (18). Sinusitis-induced subdural empyemas may not become apparent on CT scan until 1 or 2 weeks after symptoms develop, hence they may be missed on initial CT scans (306). Enhancement of membranes with gadolinium on MRI can occur when no enhancement is seen on CT (307). Diffusion MR imaging can be valuable in distinguishing subdural empyema from effusion and in the follow-up of subdural collections (308). Early diagnosis and prompt chemotherapy and surgical drainage are imperative (269). Craniotomy is the surgical procedure of choice, but more limited procedures (burr holes and craniectomies) should be performed in patients in septic shock, for patients with parafalcine empyemas, or for children with subdural empyemas secondary to meningitis (309,310). Subdural infections are caused by the same spectrum and distribution of bacteria as those causing brain abscesses (269).
Epidural Abscesses
Epidural abscesses can develop from contiguous infection of structures that surround the brain and spinal cord. They can arise from local infection or can be secondary to congenital anomalies such as dermal sinuses (311,312). Bacteremia and trauma also have been implicated. Approximately one-third of cases involve staphylococcal organisms (313). Epidural abscesses complicating sinus infections most commonly present with purulent nasal or aural discharge, fever, headache, scalp swelling, signs or symptoms of raised intracranial pressure, and, rarely, focal neurologic findings or seizures (314,315). Epidural abscesses can have a more indolent course than subdural empyemas (316).
Infection of the spinal epidural space, though extremely rare in childhood, produces a devastating neurologic picture. The main determinant of outcome for patients with spinal epidural abscess is neurologic status at the time of diagnosis (317). The infection is limited to the dorsal surface of the cord except in the cord’s lower sacral portion, where the abscess completely surrounds the cord. The anatomy of the epidural space limits extension to vertical spread and produces extradural compression. Epidural infections can result from the use of epidural catheters for prolonged postoperative analgesia in children (318).
The clinical course can be acute and rapidly progressive or chronic. The acute form, more common in children, is usually the result of a metastatic infection, whereas chronic lesions are commonly caused by a direct extension of a spinal osteomyelitis. The classic case of spinal epidural abscess follows a fairly typical pattern of development. Within 1 to 2 weeks of an infection, backache develops; this backache is enhanced by jarring or straining and is accompanied by local spinal tenderness. Within a few days, tenderness progresses to radicular pain followed by symptoms of cord involvement, including weakness of voluntary movements and impaired sphincter control. Paraplegia can be complete within a few hours or days.
In many patients, the history and neurologic signs are impossible to differentiate from those of acute transverse myelitis. Intradural extramedullary tumors can produce a similar clinical picture.
A history compatible with infection, in association with focal pain in the region of the spine, especially when accompanied by neurologic symptoms of spinal cord or root involvement, should suggest the diagnosis, which can be confirmed by neuroimaging studies (18). Prompt blood cultures can reveal the causative organism. Lumbar puncture should not be performed because of the risk of spreading the infection to the subdural or subarachnoid space. Aspiration can be performed with a large-gauge needle, but only under radiographic guidance. If the CSF is examined, it shows pleocytosis and an elevated protein level.
An epidural abscess that has produced neurologic deficit is treated by laminectomy with decompression and drainage. In view of the rapid progression of potentially irreversible symptoms, this condition is a surgical emergency. Antibiotic therapy is secondary. If tuberculosis of the spine is suspected, antituberculous therapy should be
instituted pending identification of the causative organisms. Sinus-related intracranial epidural abscesses in children may be managed without neurosurgical procedures in the setting of adequate sinus drainage, appropriate antibiotic therapy, and minimal extradural mass effect from the abscess (308).
instituted pending identification of the causative organisms. Sinus-related intracranial epidural abscesses in children may be managed without neurosurgical procedures in the setting of adequate sinus drainage, appropriate antibiotic therapy, and minimal extradural mass effect from the abscess (308).
 FIGURE 7.5. Septic discitis. Magnetic resonance imaging scan of the lumbar spine of a 14-month-old child who stopped cruising. A: Sagittal T2-weighted scan showing reduced signal in the L5/S1 intervertebral disc (arrow) and high signal in the end plate and adjacent medullary bone. B: Axial T1-weighted scan after contrast demonstrated intense contrast enhancement of the disc. C: Axial T1-weighted scan showing nerve roots surrounded by fat. (Courtesy of Dr. Philip Anslow, Department of Radiology, Radcliffe Infirmary, Oxford, England.) |
Intramedullary Abscesses
Approximately 27% of intramedullary abscesses occur before age 10 years, and 40% occur during the first two decades of life (319). The abscess develops by spread from infected contiguous dermal sinuses or, less often, from suppurative foci at other sites. In some instances, particularly since the advent of antibiotics, the disease can be insidious, and in approximately one-fourth of cases, the infection has been present for at least 4 months before diagnosis. The thoracic cord is involved in most instances. Symptoms generally simulate an intramedullary tumor.
Infections of Intervertebral Disc Space
Self-limited intervertebral disc space infections can occur in children. These infections can be caused by S. aureus or diphtheroids, but often no organism can be isolated.
Infections of the disc space are characterized by back pain with or without fever, a variable incidence of hip pain or abdominal pain, radiographic evidence of narrowing of the intervertebral space, enhanced radionuclide uptake, and an elevated sedimentation rate (320). Cross-sectional MRI is probably the most useful diagnostic procedure (321) (Fig. 7.5). Treatment by immobilization relieves pain. Antibiotics have been used, but are not always needed. Scoliosis and complete interbody ankylosis can occur (322,323). In a follow-up of 35 children on average 17 years after discitis, flexion of the low back was normal in 90% but extension was markedly restricted in 86%, 80% had narrowing of the vertebral canal, 74% had a block vertebrae, and 43% still complained of backache (324). Nonspecific discitis without bacterial cause can occur in young children. It is characterized by restriction of spinal mobility and an elevated sedimentation rate. Biopsy usually reveals chronic or subacute inflammation, although approximately one-third of the biopsies yield normal tissue. The disease is usually self-limiting in most cases with only minor residual radiographic changes (325).
Infections of the disc space are characterized by back pain with or without fever, a variable incidence of hip pain or abdominal pain, radiographic evidence of narrowing of the intervertebral space, enhanced radionuclide uptake, and an elevated sedimentation rate (320). Cross-sectional MRI is probably the most useful diagnostic procedure (321) (Fig. 7.5). Treatment by immobilization relieves pain. Antibiotics have been used, but are not always needed. Scoliosis and complete interbody ankylosis can occur (322,323). In a follow-up of 35 children on average 17 years after discitis, flexion of the low back was normal in 90% but extension was markedly restricted in 86%, 80% had narrowing of the vertebral canal, 74% had a block vertebrae, and 43% still complained of backache (324). Nonspecific discitis without bacterial cause can occur in young children. It is characterized by restriction of spinal mobility and an elevated sedimentation rate. Biopsy usually reveals chronic or subacute inflammation, although approximately one-third of the biopsies yield normal tissue. The disease is usually self-limiting in most cases with only minor residual radiographic changes (325).
Intraventricular Shunt Infections
The management of infected cerebrospinal shunts is covered in Chapter 5.
CONGENITAL INFECTIONS
Introduction
Several pathogens, grouped traditionally as TORCH infections—an acronym designating toxoplasmosis, others, rubella, cytomegalovirus, and herpes (326)—are potential causes of deafness, vision loss, and behavioral or neurologic disorders among young children throughout the world (Table 7.10). Although the introduction of the rubella vaccine in the late 1960s led to a dramatic reduction in the incidence of the congenital rubella syndrome (CRS), the last several decades have brought only modest changes in the epidemiology of congenital infections. This section describes the major pathogens causing intrauterine infections and summarizes information on pathogenesis, clinical manifestations, treatment, and prevention.
TABLE 7.10 Infectious Agents Potentially Associated with Intrauterine Central Nervous System Infections | |
|---|---|
|
Pathogenesis and Pathology
Intrauterine infections reflect the complex interaction of the mother’s immune status, the placenta, the fetus, and the infectious pathogen (327). Relatively few microorganisms can cross the placenta and damage the developing fetus. Women who possess antibodies to specific pathogens have reduced risks of transmitting the agents to their infants. By contrast, women who experience primary infections with these pathogens during their pregnancies have substantial risks of delivering infants with symptomatic congenital infections.
The agents causing intrauterine infections reach the placenta and the fetus during maternal viremia, parasitemia, or spirochetemia. There, the pathogens replicate, invade the fetal circulation, and disseminate hematogenously to the target organs of the developing fetus. For certain agents, such as rubella and varicella-zoster virus (VZV), the fetal consequences of infection reflect the timing of maternal infection (328,329). Maternal rubella during the initial 8 weeks, for example, produces cataracts and congenital heart lesions, whereas sensorineural hearing loss correlates with infection during the first 16 weeks. Stillbirth or spontaneous abortion can also occur during this time interval. The risk of the fetal varicella syndrome is restricted to first-trimester maternal varicella.
During maternal infections with Toxoplasma gondii the likelihood of transmission to the fetus increases during gestation, but maternal infections early in pregnancy are more likely to produce several fetal infections (330). For other agents, such as cytomegalovirus (CMV), the relationship between the timing of infection and the likelihood of fetal damage is less clear (331,332). CMV infection during critical stages of fetal development can produce severe CNS malformations, such as lissencephaly or schizencephaly (333,334), but damage to other tissues, including the retina, cochlea, liver, or spleen, can occur irrespective of the timing of maternal infection.
Intracranial calcifications, a hallmark of intrauterine infections, accompany infections with several pathogens, including CMV, rubella, herpes simplex virus (HSV), VZV, human immunodeficiency virus (HIV), lymphocytic choriomeningitis (LCM) virus, and T. gondii. Calcifications indicate tissue necrosis and dystrophic changes. The location of the calcifications provides a useful clue regarding the etiology of intrauterine infections. Periventricular calcifications commonly occur with CMV (Fig. 7.6), rubella, and LCM virus, whereas diffuse parenchymal as well as periventricular calcifications suggest congenital
toxoplasmosis (Fig. 7.7). By contrast, calcifications associated with HSV, VZV, and HIV tend to involve the thalamus and basal ganglia.
toxoplasmosis (Fig. 7.7). By contrast, calcifications associated with HSV, VZV, and HIV tend to involve the thalamus and basal ganglia.
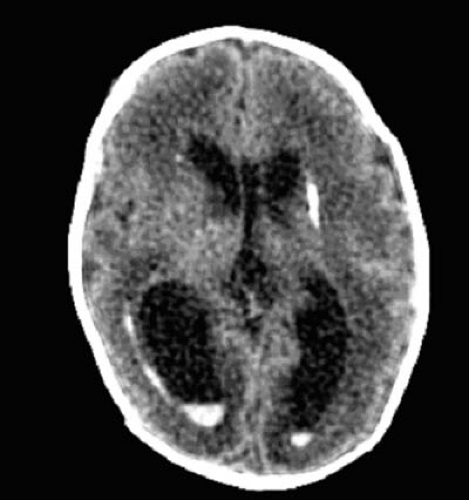 FIGURE 7.6. Unenhanced head CT of a premature infant with congenital cytomegalovirus infection showing a dense periventricular calcification as well as an intraventricular hemorrhage. |
Cystic necrosis occurs during intrauterine infections with HSV and VZV as well as with Venezuelan equine encephalitis virus, a pathogen endemic to Latin America and a rare cause of intrauterine infection. These pathologic abnormalities result from tissue invasion, virus replication, and immune-mediated tissue destruction. In severe cases, tissue destruction can lead to extensive encephalomalacia or hydranencephaly. Infectious vasculopathy contributes to the tissue changes associated with several intrauterine infections, including rubella, congenital syphilis, CMV, and VZV. The prevention and treatment of the various congenital infections are summarized in Table 7.11.
Cytomegalovirus (CMV)
Epidemiology
In the United States and many developed countries, 0.4% to 2.5% of newborns shed CMV at birth (331). The incidence of acquired CMV infection among children and adults, including women of childbearing age, averages 1% to 2% per year. By adulthood, 40% to 100% of the population has been infected with CMV. Infants acquire CMV postnatally from their breast-feeding mothers, and young children become infected through contact with CMV-excreting playmates (335). Adults acquire CMV via blood transfusion, sexual contact, organ transplantation, or contact with young children (336). Adults with multiple sexual partners have increased rates of CMV acquisition (332).
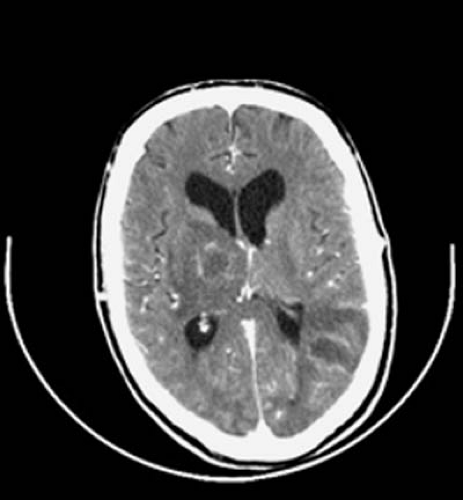 FIGURE 7.7. Unenhanced head CT of a child with congenital toxoplasmosis showing mild cerebral atrophy and scattered, parenchymal calcifications. |
Pathogenesis and Pathology
Approximately 40% of women who acquire primary CMV infection during pregnancy transmit CMV to the fetuses, and 5% to 10% of the infected infants have CMV disease (337). Although seronegative women have the greatest risk of transmitting CMV to their unborn infants, fetal infections can occur in CMV-seropositive women who are reinfected with new CMV strains (338). CMV infection begins with replication of the virus in lymphoid tissues adjacent to the genital tract or oropharynx. Viremia then develops and leads to infection of systemic organs, including the placenta of pregnant women. Although most fetal tissues support CMV replication, major sites of infection are the liver, spleen, brain, and retina. The virus enters the brain via the choroid plexus and spreads centrifugally from the subependymal region. Pathologic changes within the brain include hemorrhage, cystic degeneration, dystrophic calcification, and perivascular mononuclear inflammation (332).
TABLE 7.11 Prevention and Treatment of Intrauterine Infections | |||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||
Clinical Manifestations
The majority of CMV-infected neonates have no signs of CMV infection at birth. Approximately 5% to 10% of the infected newborns have systemic signs (currently designated “congenital CMV disease”) (Table 7.12) consisting of jaundice, hepatomegaly, splenomegaly, petechial or purpuric rash, intrauterine growth retardation, or respiratory distress (339,340,341,342,343). Neurologic features include microcephaly, chorioretinitis, seizures, sensorineural hearing loss, and alterations in muscle tone.
Diagnosis
The diagnosis of intrauterine infection is established by detecting CMV in clinical samples during the first 3 weeks of life (331,332). Because congenitally infected infants shed enormous quantities of CMV in their urine, culturing urine or saliva using the shell vial assay is the gold standard for the diagnosis of intrauterine CMV infection. Performing PCR or assaying CMV-specific IgG or IgM in the infant’s serum has much lower sensitivity. CMV antigenemia assays have no role in the diagnosis of intrauterine CMV infections.
Approximately 50% of the infants with congenital CMV disease have periventricular calcifications (Fig. 7.6), and infants with calcifications have higher rates of permanent neurodevelopmental disabilities (344,345). Neuroimaging studies may also show periventricular leukomalacia, polymicrogyria, pachygyria, schizencephaly, or lissencephaly. Because CT detects small calcifications more effectively than MRI, CT is the preferable imaging study in the perinatal period. MRI provides more accurate
information regarding cortical dysplasia, polymicrogyria, and other disorders of neuronal migration associated with congenital CMV infection.
information regarding cortical dysplasia, polymicrogyria, and other disorders of neuronal migration associated with congenital CMV infection.
TABLE 7.12 Neonatal Clinical Features in Infants with Proven Intrauterine Infections with Cytomegalovirus and Toxoplasma gondii, the Most Frequent Pathogens Associated with Congenital Infections in Most Developed Nations | ||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
Prevention, Treatment, and Prognosis
There are no vaccines for preventing CMV infections. Pregnant women can reduce their risk of infection by avoiding direct contact with the urine and saliva of young, toddler-aged children (346). Should inadvertent skin contact occur, hand washing with soap and water eliminates the virus from the skin. Postnatal therapy with ganciclovir can reduce the severity of sensorineural hearing loss and can treat CMV-induced pneumonia in congenitally infected infants (341). In the treatment trial sponsored by the National Institute of Allergy and Infectious Diseases–Children’s Antiviral Study Group (NIH-CASG), infants received 6 mg/kg per dose intravenously every 12 hours for 6 weeks (Table 7.11) (341,347).
The majority of infants with silent CMV infections escape neurodevelopmental sequelae, but 7% to 10% of these will later exhibit sensorineural hearing loss. Because of the high incidence of intrauterine CMV infection, this disorder represents the most common cause of nongenetic deafness in many regions (348). By contrast, infants with congenital CMV disease have high rates (greater than 90%) of neurodevelopmental sequelae, consisting of cerebral palsy, seizures, sensorineural hearing loss, mental retardation, behavioral disorders, and vision loss (331,332,349). Approximately 9,000 infants annually in the United States have audiologic or neurologic sequelae of intrauterine CMV infections (331).
Toxoplasma gondii
Epidemiology
Humans acquire T. gondii by ingesting undercooked meat that contains bradyzoites, the encysted form found in tissues, or by ingesting fruits, vegetables, and other foods contaminated by oocysts, the highly infectious form of the organism (350). Infected cats, major contributors to human infection, excrete vast quantities of oocysts (351). Approximately 0.1% to 2% of the adult population acquires T. gondii annually. Rates of congenital infection vary from 1 per 12,000 live births to as high as 1 per 1,000 (330,347,350,351,352).
Pathogenesis and Pathology
Fetal infections complicate approximately 40% of the T. gondii infections in pregnant women (330,350). Healthy women infected with T. gondii rarely have recognizable symptoms, although an acute mononucleosis-like illness develops occasionally. Infections during the first trimester, when transmission to the fetus is less likely, can produce spontaneous abortion, whereas infections during the third trimester, when transmission frequently occurs, usually cause subclinical fetal infection. Infections during the second trimester often produce severe fetal disease. Congenital toxoplasmosis produces a diffuse, granulomatous meningoencephalitis that can lead to aqueductal obstruction, fetal hydrocephalus, and dystrophic parenchymal calcification.
Clinical Manifestations
Approximately 85% of the infants with intrauterine T. gondii infections lack symptoms at birth, but many of these infants later have ophthalmologic or neurologic sequelae (330,342,350,353). Symptomatic neonates exhibit jaundice, splenomegaly, hepatomegaly, fever, anemia, chorioretinitis, hydrocephalus or microcephaly, and petechiae, secondary to thrombocytopenia (Table 7.12; Fig. 7.8) (330). Intrauterine coinfections with T. gondii and HIV or CMV can occur.
Diagnosis
The diagnosis of intrauterine infection with T. gondii can be made postnatally by detecting anti-Toxoplasma antibodies of several classes—IgM, IgG, IgA, and IgE—in the newborn infant’s serum (354,355). Serologic studies of infants
and their mothers should be sent to an experienced laboratory, such as the Palo Alto Laboratories (phone number 650-853-4828), that performs a battery of serologic studies for T. gondii. As many as 25% of infected infants lack anti-Toxoplasma IgM. Inoculation of fresh placental tissues into laboratory mice can be used to confirm the diagnosis of congenital toxoplasmosis, and PCR can detect T. gondii in clinical specimens (356). Imaging studies in infants with congenital toxoplasmosis may show intracranial calcifications and ventriculomegaly, reflecting hydrocephalus or passive ventricular enlargement (330).
and their mothers should be sent to an experienced laboratory, such as the Palo Alto Laboratories (phone number 650-853-4828), that performs a battery of serologic studies for T. gondii. As many as 25% of infected infants lack anti-Toxoplasma IgM. Inoculation of fresh placental tissues into laboratory mice can be used to confirm the diagnosis of congenital toxoplasmosis, and PCR can detect T. gondii in clinical specimens (356). Imaging studies in infants with congenital toxoplasmosis may show intracranial calcifications and ventriculomegaly, reflecting hydrocephalus or passive ventricular enlargement (330).
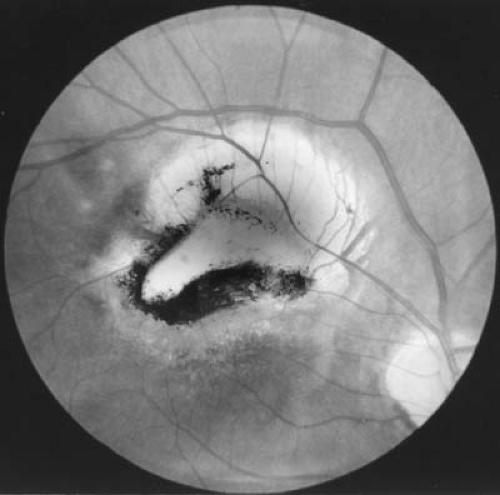 FIGURE 7.8. Chorioretinitis in toxoplasmosis. (Courtesy of Dr. John Chang, Department of Ophthalmology, University of California, Los Angeles, CA.) |
Prevention, Treatment, and Prognosis
Pregnant women can reduce their risk of exposure to T. gondii by avoiding cat excreta and modifying their dietary habits (357). Treating infected mothers with spiramycin or pyrimethamine-sulfadiazine has been advocated to reduce the risk of congenital toxoplasmosis, although the effectiveness of these strategies is unproven (358,359). Therapeutic abortion has been considered when the diagnosis of intrauterine toxoplasmosis is confirmed and CNS damage has been detected by fetal ultrasonography.
The neurologic prognosis of infants with congenital toxoplasmosis can be improved by early shunting of obstructive hydrocephalus and prolonged antitoxoplasma therapy postnatally (330,343,360). Infants should receive sulfadiazine, 100 mg/kg per day, and pyrimethamine, 1 mg/kg per day, for 6 months followed by the same dose three times per week for an additional 6 months. Infants require folinic acid, 5 to 10 mg three times weekly. Among infants who do not receive antitoxoplasma therapy, 80% have epilepsy, 70% have cerebral palsy, 60% have visual impairment, and nearly 60% have intelligence quotients less than 70 (330). Treated infants have considerably lower rates of neurodevelopmental complications (343,360).
Herpes Simplex Virus Type 2
Epidemiology
Intrauterine HSV disease, a rare disorder, nearly always results from HSV-2 infection. The incidence of HSV-2 infection is highest among young women, approximately 2% of whom acquire the virus each year (361); approximately 30% of 30-year-old women have serologic evidence of prior HSV-2 infection. The risk of HSV-2 infection among U.S. women is associated with several sociodemographic factors, including African-American race, lower income, greater number of lifetime sexual partners, earlier age of sexual intercourse, and drug use (362).
Pathogenesis and Pathology
As many as 2% of pregnant women become infected with HSV during pregnancy, but the majority of these infections do not produce fetal or neonatal disease (363). Primary maternal infection usually occurs without recognizable symptoms. The risk of intrauterine transmission of HSV-2 to the fetus appears to be very low. Fewer than 50 cases of intrauterine HSV infection have been described. HSV may reach the fetus during maternal viremia or through an ascending intrauterine infection. The virus replicates in several tissues, including the skin, liver, lung, eye, and brain, producing tissue necrosis, cystic degeneration, and dystrophic calcifications (364).
Clinical Manifestations
Infants with congenital HSV infections have skin vesicles or scarring, chorioretinitis, microcephaly, and microophthalmia (365,366,367). Intrauterine growth retardation can also be present. Of 13 infants identified by the NIH-CASG, 12 had skin lesions, 8 had chorioretinitis, 7 had microcephaly, and 5 had hydranencephaly, evidence of severe brain necrosis (366). Less severe congenital HSV-2 infections likely occur.
Diagnosis
The diagnosis of congenital HSV-2 infection can be made by detecting the virus in skin lesions or body fluids, using cell culture or PCR (368). Samples for virus isolation should include skin vesicle fluid, CSF, and swabs of the conjunctiva, rectum, and oropharynx. Ophthalmologic examination may reveal chorioretinitis and microphthalmia. Infants with intrauterine HSV infections frequently have dense calcifications of the basal ganglia and thalami, diffuse, hemispheric cystic lesions, and a lissencephalic-like cerebral cortex (369).
Prevention, Treatment, and Prognosis
Preventing neonatal HSV-2 infection, including intrauterine infection, remains problematic, and most women who give birth to infants with congenital HSV infection have no history of genital HSV infection (369). Although infants with neonatal HSV infections require prolonged high-dose therapy with acyclovir (60 mg/kg per day for 21 days) (369), acyclovir has little or no benefit for the severe CNS damage that accompanies intrauterine HSV-2 infection. Infants with congenital HSV-2 infections often die in infancy; survivors usually have cerebral palsy, epilepsy, and mental retardation (366).
Varicella-Zoster Virus
Epidemiology
Chickenpox affects approximately 0.05% of pregnant women (370). Women who acquire VZV before week 20 of gestation have a 2% risk of delivering an infant with the fetal varicella syndrome (371,372). By contrast, fetal varicella syndrome is highly unlikely in women who have chickenpox during the last half of pregnancy. Maternal VZV reactivation (zoster) poses little or no risk to the developing fetus.
Pathogenesis and Pathology
VZV displays tropism for the brain, retina, skin, and long tracts or neurons of the spinal cord. Lytic infection of neurons depletes neuronal populations, leading to the clinical manifestations described in the following paragraph. Pathologic changes include tissue necrosis, mononuclear inflammation, and cystic degeneration. CNS necrosis can manifest as severe cystic encephalomalacia or hydranencephaly (364).
Clinical Manifestations
The clinical manifestations of the congenital varicella syndrome consist of intrauterine growth retardation, limb hypoplasia, chorioretinitis, cataracts, microphthalmia, cutaneous scaring (cicatrix), and neurologic abnormalities (329,364,373,374). The latter can include microcephaly, hydrocephalus/hydranencephaly, seizures, Horner syndrome, and cranial neuropathies.
Diagnosis
Infection can be established by detecting the virus in fetal tissues using culture or PCR and by detecting VZV-specific IgM in fetal blood samples obtained by cordocentesis (376,376). At birth, infants with the fetal varicella virus syndrome no longer shed VZV, and VZV-specific IgM can be undetectable. Imaging studies may show intracranial calcifications, cortical dysplasia, or hydranencephaly (364,377).
Prevention, Treatment, and Prognosis
Although the efficacy of varicella-zoster immune globulin (VZIG) in preventing the fetal varicella syndrome is unproven, VZIG should be considered when susceptible pregnant women have been exposed to persons with chickenpox (378). VZIG should be given within 96 hours of the exposure. Postnatal therapy with acyclovir or other antiviral medications provides no benefit for infants with the congenital varicella syndrome. A substantial number of infants with the fetal varicella syndrome will die in infancy, and survivors usually have permanent sequelae affecting vision or neurologic development (329,364).
Lymphocytic Choriomeningitis Virus
Epidemiology
Lymphocytic choriomeningitis (LCM) virus, an arenavirus that naturally infects the feral house mouse, Mus musculus, can also infect humans (379). Seroprevalence rates of 0.3% to 5.0% have been observed among persons living in the United States, Canada, and Argentina (380,381,382,383). Intrauterine LCM virus infection is presumed to be a rare condition, but no surveillance studies have been performed to establish the incidence of the disorder.
Pathogenesis and Pathology
Human infection results from inhalation of infected aerosols, direct contact with LCM virus-shedding animals, or ingestion of material contaminated by infected excreta (384). Infected adults can experience an influenza-like illness with fever, malaise, and myalgia. Fetal infections are associated with periventricular calcifications, cortical dysplasia, and diffuse lymphocytic meningitis that can involve the ependyma of the aqueduct of Sylvius and cause hydrocephalus (385).
Clinical Manifestations
The clinical features of congenital LCM virus infection mimic those of intrauterine CMV disease and congenital toxoplasmosis (379,385,386). Among 26 reported cases reviewed by Wright and colleagues in 1997, 88% had chorioretinopathy, 43% had macrocephaly at birth, and 13% were microcephalic (385). Occasional infants had vesicular or bullous skin lesions. Virtually all infants were born at term and had normal birth weights. Approximately 50% of mothers who gave birth to the infants reviewed in this study recalled influenza-like illness during pregnancy, and one-fourth reported contact with rodents, usually mice, during their pregnancy.
Diagnosis
The diagnosis of congenital LCM virus infection is established by serologic studies of the mother and infant (379,385). Antibodies against LCM virus can be detected in the infant’s serum or CSF. Because of the low seroprevalence of LCM virus infection in the general population, detecting LCM virus-specific antibodies, particularly LCM virus-specific IgM, strongly supports the diagnosis. The virus can be detected by PCR, but this method does not yet
have diagnostic utility in human infections. Imaging studies in infants with congenital LCM virus infections may show hydrocephalus or cortical dysplasia (385).
have diagnostic utility in human infections. Imaging studies in infants with congenital LCM virus infections may show hydrocephalus or cortical dysplasia (385).
Prevention, Treatment, and Prognosis
No vaccine is available for preventing LCM virus infections; pregnant women should avoid mice and pet hamsters during their pregnancy. Effective postnatal antiviral therapy of LCM virus infections has not been described. Approximately 35% of the reported infants died from complications of congenital LCM virus infection (379,385). Surviving infants commonly had cerebral palsy, epilepsy, vision loss, and mental retardation. LCM virus-infected infants with hydrocephalus may require shunt placement.
Rubella
Epidemiology
In the pre-vaccine era, epidemics of rubella caused thousands of cases of the congenital rubella syndrome (CRS), but after licensure of the rubella vaccine in 1969 and aggressive immunization programs in the 1980s, the incidence of the CRS in the United States and other nations declined dramatically (386). In the United States, only one to three cases of CRS were reported to the Centers for Disease Control and Prevention annually by the late 1980s, an incidence of less than 0.1 case of CRS per 100,000 live births (387). In recent years, CRS in the United States has virtually disappeared except among the offspring of immigrants from regions without compulsory immunization (388).
Pathogenesis and Pathology
Maternal rubella during the initial 8 weeks produces cataracts and congenital heart lesions, whereas hearing loss correlates with infection during the first 16 weeks (328). Infections after the week 16 of gestation usually leave no sequelae other than a risk of sensorineural hearing loss. Pathologic changes within the CNS consist of diffuse meningoencephalitis, cystic degeneration, and dystrophic calcification. Cardiac lesions include peripheral pulmonic stenosis, patent ductus arteriosus, and atrial or ventricular septal defects (389).
Clinical Manifestations
Infants with CRS can have cataracts, retinopathy, microphthalmia, microcephaly or sensorineural hearing loss, meningoencephalitis, osteopathy, pneumonitis, hepatitis, hepatosplenomegaly, thrombocytopenia, and jaundice (Table 7.12) (389,390,391). The propensity of the rubella virus to infect the heart causes myocarditis, patent ductus arteriosus, valvular stenosis, or septal defects at the atrial or ventricular level (389).
Diagnosis
CRS is diagnosed by isolating virus from nasal secretions, urine, or CSF or detecting rubella virus-specific IgM in the infant’s serum (370,392). Postnatal persistence of rubella-specific IgG supports the diagnosis of CRS. Imaging studies can show periventricular calcifications, periventricular leukomalacia, or subependymal cysts.
Prevention, Treatment, and Prognosis
CRS cannot be treated effectively by postnatal antiviral therapy; infection must be prevented by vaccination. Maternal antibody prior to conception or at the time of maternal rubella exposure indicates that the fetus is not at risk. Termination of pregnancy is considered in confirmed primary rubella infections early in gestation (370). When termination is not an option, immune globulin can be considered, although the efficacy of this approach is unproven (370). Progressive sensorineural hearing loss can develop in children who survive CRS, and children with CRS also have an increased risk of growth failure or diabetes mellitus in the second or third decade of life (393,394,395). Neurologic sequelae include microcephaly, language delay, autistic features, and developmental or mental retardation.
Syphilis
Epidemiology
The overall incidence of congenital syphilis is low in the United States, but Treponema pallidum remains endemic in large urban areas and the rural South (396,397,398,399). In virtually all other developed countries syphilis rarely occurs. A transient resurgence of congenital syphilis in the United States in the late 1980s and early 1990s was attributed to maternal use of illicit drugs, especially cocaine (398).
Pathogenesis and Pathology
Untreated early maternal infection causes perinatal death, stillbirth, or miscarriage in approximately 40% of pregnancies (400). Although congenital syphilis can occur during all stages of maternal infection, fetal infection is most likely during secondary-stage maternal infections, when rates of transmission range from 60% to 100%. Maternal–fetal transmission complicates approximately 30% of latent maternal infections. Pathologic features in infants with congenital syphilis include periostitis, osteochondritis, leptomeningitis, endarteritis, periarteritis, and mixed
inflammatory infiltration of multiple organs or tissues, including the liver, lungs, skin, eyes, inner ear, and central nervous system (401).
inflammatory infiltration of multiple organs or tissues, including the liver, lungs, skin, eyes, inner ear, and central nervous system (401).
Clinical Manifestations
Early signs of congenital syphilis, present at birth or evident during the first 2 years of life (399,400,401), consist of intrauterine growth retardation, rash, hepatosplenomegaly, jaundice, lymphadenopathy, pseudoparalysis, and bony abnormalities. Hemolytic anemia, thrombocytopenia, leukocytosis, or leukopenia can be present, and long-bone radiographs in the perinatal period may show osteochondritis. The latter can mimic nonaccidental trauma. Late signs of congenital syphilis, features evident after 2 years of age, include sensorineural deafness, dental abnormalities, saddle nose, saber shins, hydrocephalus, and developmental delay (400,401).
Diagnosis
Infants with suspected congenital syphilis require (a) quantitative treponemal and nontreponemal assays of serum, (b) routine studies and nontreponemal and treponemal assays of CSF, and (c) long-bone radiographs. Definitive diagnosis is made by demonstrating spirochetes in exudates or tissues using dark-field or direct immunofluorescence microscopy (402). Serologic tests consist of nontreponemal tests, such as the Venereal Disease Research Laboratory (VDRL) slide test or rapid plasmin reagin (RPR), and treponemal tests, such as the fluorescent treponemal antibody absorption (FTA-ABS) test or the T. pallidum particle agglutination (TP-PA). Infants likely have congenital syphilis when spirochetes are present in tissues or when they have reactive CSF VDRL or FTA-ABS tests or serum quantitative treponemal tests four times higher than the mother (402).
Prevention and Treatment
Pregnant women with acquired syphilis require penicillin treatment, using one to three doses of penicillin G benzathine (2.4 million units intramuscularly), at 1-week intervals if more than one dose is given, irrespective of the timing of pregnancy (404). No effective alternatives exist for penicillin-allergic persons, so desensitization should be performed. Neonates with proven or highly suspected symptomatic congenital syphilis require aqueous crystalline penicillin G, 50,000 U/kg intravenously every 12 hours during the first week of life and every 8 hours thereafter for a total of 10 days (401). Alternatively, procaine penicillin G can be given intramuscularly at a dose of 50,000 U/kg once a day for 10 days. If penicillin G cannot be given, treatment recommendations can be found at http://www.cdc.gov/nchstp/dstd/penicillinG.htm. Infectious disease experts should be consulted regarding current treatment strategies for infants whose mothers received inadequate treatment, infants with asymptomatic infections, or infants older than 4 weeks with possible syphilis and neurologic involvement.
Congenital Chagas Disease
Infants with congenital Chagas disease, caused by intrauterine infection with Trypanosoma cruzi, resemble those with congenital toxoplasmosis (403,404). Hepatosplenomegaly, petechiae, jaundice, anemia, and seizures are potential complications. The diagnosis can be established by detecting serologic responses or parasitemia. Infected persons, including infants, can be treated with nifurtimox (405).
VIRAL INFECTIONS
Viral infections of the CNS are usually benign and self-limited with a good clinical outcome. A subset of viruses, however, can result in a severe clinical course with a high morbidity and mortality. Acute encephalitis is more common in children (more than 16 cases per 100,000 patient-years) than in adults (between 3.5 and 7.4 cases per 100,000 patient-years) (406). The number of annual cases of primary and postinfectious encephalitis reported to the Centers for Disease Control and Prevention from 1989 to 1994 ranged from 717 to 1,341 and from 82 to 170, respectively. Since 1995, these clinical entities are no longer nationally notifiable.
Pathogenesis
Viruses can cause CNS damage through direct invasion of central nervous tissues (primary or infectious encephalitis) or through the generation of an immune response to a systemic viral infection (postinfectious encephalomyelitis) (407).
In a naturally acquired infection, virus spreads to the CNS through two main mechanisms: hematogeneous and neuronal. These two mechanisms are not mutually exclusive and can occur concurrently to spread a virus.
Hematogenous Spread
The most common pathway for spread of virus to the CNS is by way of the bloodstream. Viral growth generally begins in extraneural tissue: poliomyelitis in the gut or regional lymphatics; herpes simplex in the respiratory tract or gastrointestinal mucosa; arbovirus in the vascular epithelium; and coxsackievirus, reovirus, rabies, and variola in brown fat. The viremia is maintained by shedding of the virus, absorption into red blood cells, or growth within
leukocytes. Virus in blood can enter the CSF by either passing through or growing through the choroid plexus. Growth of virus in the choroid plexus might explain the ease with which echovirus and coxsackievirus can be isolated from the CSF. Migration of infected phagocytes through cerebral vessels and replication of virus in the endothelial cells with growth or passive transfer through the blood–brain barrier also can occur.
leukocytes. Virus in blood can enter the CSF by either passing through or growing through the choroid plexus. Growth of virus in the choroid plexus might explain the ease with which echovirus and coxsackievirus can be isolated from the CSF. Migration of infected phagocytes through cerebral vessels and replication of virus in the endothelial cells with growth or passive transfer through the blood–brain barrier also can occur.
Once within the nervous system, the virus can spread by direct contiguity or through the limited extracellular space. Neurovirulent viruses that lack the neuroinvasive ability to cross the blood–brain barrier are able to produce infection once the barrier is breached by bacterial lipopolysaccharide (407).
Neuronal Spread
Less commonly, neuronal spread occurs from peripheral and cranial nerves. Viruses can enter the nervous system tissue by centripetal spread along the endoneurium, perineural Schwann cells, and fibrocytes of peripheral nerves. Once the virus has gained access to the spinal cord by this route, it can spread randomly along the endoneurium or the interstitial space within the nerve. Spread within the nerve is significant for rabies and for some herpes simplex infections. Penetration at sensory or motor nerve endings with transport to specific sensory ganglion cells or motor neurons by retrograde axoplasmic flow has been demonstrated for herpes simplex, polio viruses, and rabies virus.
Spread by way of the olfactory system has been demonstrated in herpes simplex, poliomyelitis, and arbovirus infections. These viruses spread along olfactory rods and nerve fibers to the mitral cell neurons of the olfactory bulb, or along the interstitial spaces, endoneurium, and perineurium of the olfactory nerves to the parenchymal cells of the olfactory bulb, and from there along the subarachnoid cuffs to the meninges (408).
Pathology
Cells of the CNS vary in their susceptibility to viral infection. Thus, herpes simplex seems to infect all cell types, whereas poliomyelitis has a predilection for the larger motor cells, Purkinje cells, and cells of the reticular formation. Susceptibility is determined by cell receptor sites and by the route of entry.
The reactions of brain tissue to viral invasion are similar in all forms of encephalitis. The most obvious histologic lesion is a cellular infiltration. Initially, this consists largely of polymorphoneutrophils; later, round cells predominate. Proliferation of microglial cells can be diffuse or focal. With diffuse proliferation, the cells accumulate around degenerated or dying nerve cells, a phenomenon associated with neuronophagia. Most commonly, alterations within nerve cells consist of a loss of Nissl granules and eosinophilia of the cytoplasm. Inclusion bodies within neurons and glial cells, such as the Negri bodies seen in rabies, can be typical of the disease or can represent nonspecific degeneration products.
Arteritis and other alterations of the vessel walls are observed in a number of conditions, notably in the equine encephalitides. Demyelination can result from the loss of cortical nerve cells or can occur independently, as in subacute sclerosing panencephalitis (SSPE) or other slow virus infections.
Viral infections are characterized by intrathecal production of oligoclonal immunoglobulin within the CNS. Viral-specific IgM is produced during the early stage of infection, with specific IgG and IgA produced slightly later.
The patient’s humoral and cellular immune response to infection can contribute to the disease picture. For instance, in experimental lymphocytic choriomeningitis, the virus causes severe meningeal infiltrates only in immunologically mature and competent animals. TNF, the cytokine associated with inflammation in bacterial infections, is not demonstrable in many viral infections of the nervous system (409,410). Leukotriene B4, a potent chemotactic factor for leukocytes, and leukotriene C4, which increases vascular permeability and causes smooth muscle contraction, are increased in the CSF of children with aseptic meningitis and encephalitis (411). Suppression of the immune response with dexamethasone may have no benefit or can even be deleterious (412). For a review of T-cell activation, critical to activation of the immune response to viruses, see the publication by Reiser and Stadecker (413).
Apoptosis is a common pathway to virus-induced cell death. It can result from viral infection of cells or from the action of leukocytes and their products. This subject has been extensively reviewed by Razvi and Welsh (414).
Clinical Manifestations
Viruses can cause encephalitis (inflammation of the brain) and meningitis (inflammation of the meninges). These processes can be seen independently as well as concurrently.
Acute Encephalitis
The initial clinical picture can be protean, manifesting as a systemic infection without neurologic symptoms or findings. Fever, irritability, headaches, nausea, vomiting, poor appetite, and restlessness precede neurologic symptoms. In Finnish studies, the most characteristic neurologic symptoms of viral encephalitis during the acute phase were lowered consciousness manifested by disorientation, confusion, somnolence, or coma. These symptoms were recorded in 58% of patients. Ataxia was seen in 58%; an altered mental status, notable aggressiveness, and apathy were seen in 40%. Seizures, either generalized (32%), focal (5%), or both, were encountered less commonly.
Neurologic findings were generally nonspecific, and meningeal symptoms and signs were not always present (415,416). The incidence of CNS viral infections in association with neonatal seizures may be much higher than previously reported (417). The CSF examination was often frequently normal early in the disease (418). As a general rule, the initial CSF pleocytosis is characterized by a rapidly decreasing proportion of granulocytes. The transient elevation in CSF granulocyte colony-stimulating factor observed during the initial stages of aseptic meningitis is associated with this change (419). The development of PCR has greatly increased our ability to diagnose viral infections of the CNS, particularly for herpes and enteroviral infections (420). Young age, disorientation, and loss of consciousness are serious prognostic signs in encephalitis, especially if the encephalitis is caused by herpes simplex. In the study by Klein and coworkers, focal signs on neurologic examination and abnormal neuroimaging studies were the only two factors present at admission that predicted a poor short-term outcome (421). Unilateral hyperperfusion demonstrated by single photon emission CT (SPECT) also is associated with a poor prognosis in encephalitis (422).
Neurologic findings were generally nonspecific, and meningeal symptoms and signs were not always present (415,416). The incidence of CNS viral infections in association with neonatal seizures may be much higher than previously reported (417). The CSF examination was often frequently normal early in the disease (418). As a general rule, the initial CSF pleocytosis is characterized by a rapidly decreasing proportion of granulocytes. The transient elevation in CSF granulocyte colony-stimulating factor observed during the initial stages of aseptic meningitis is associated with this change (419). The development of PCR has greatly increased our ability to diagnose viral infections of the CNS, particularly for herpes and enteroviral infections (420). Young age, disorientation, and loss of consciousness are serious prognostic signs in encephalitis, especially if the encephalitis is caused by herpes simplex. In the study by Klein and coworkers, focal signs on neurologic examination and abnormal neuroimaging studies were the only two factors present at admission that predicted a poor short-term outcome (421). Unilateral hyperperfusion demonstrated by single photon emission CT (SPECT) also is associated with a poor prognosis in encephalitis (422).
Viral Meningitis (Aseptic Meningitis)
This is a clinical syndrome characterized in children by fever, abrupt onset of headaches, vomiting, irritability, and physical findings of meningeal irritation such as Kernig and Brudzinski signs. Infants present with a mild syndrome without the physical findings found in older children. The spinal fluid reveals less than 1,000 white blood cells with a mononuclear predominance, although early in the course of the illness, a polymorphonuclear predominance may be observed. CSF glucose is normal or slightly decreased, and CSF protein is slightly elevated. The Gram stain is universally negative, as are bacterial, mycobacterial, and fungal cultures.
Diagnosis
The diagnosis of a presumptive CNS viral infection starts with a thorough and meticulous history looking for significant exposures within the previous 2 to 3 weeks. The exposure of the patient to sick humans or animals (especially horses), travel history, and exposure to mosquitoes, rodents, and ticks helps to ascertain potential etiologies. The physical examination provides useful information about the extent of the disease and guides the choice of diagnostic studies required.
The analysis of the CSF is crucial in the search of the etiologic diagnosis. The cytochemical analysis combined with specific virologic (PCR and cultures) and serologic tests (specific IgM, IgG, or both) can assist in confirming the etiology. CSF also helps to rule out other treatable infectious or noninfectious causes, which could mimic a viral process. Thus, it is important in a child with a clinical picture of aseptic meningoencephalitis to rule out fungal, mycobacterial, and parasitic infections by performing appropriate stains and cultures. Bacterial infections of the CNS need to be ruled out by Gram stains, rapid antigen tests, and cultures. Viral cultures of blood, stool, and throat serve as a useful diagnostic tool. Blood obtained in the beginning of the clinical course as well as 2 to 3 weeks later can be used for the serologic confirmation of diseases through the observation of a fourfold increase in the specific antibody titer.
The value of neuroimaging studies lies not in determining the etiology of the viral encephalitis or meningitis, but in assessing the degree of CNS damage. For this purpose, MRI has been found to be more helpful than CT (423).
By the combined use of the various diagnostic studies, the etiology of viral meningitis or encephalitis can be determined in approximately 60% of patients (416).
This section considers the various types of viral infections according to etiology. A survey of their relative frequencies, as derived from data of Koskiniemi and Vaheri, is presented in Table 7.13 (424). Table 7.14 compiles the etiologic agents, type of disease, mechanism of acquisition,
frequency, fatality rates, and laboratory confirmation, as derived from data from Cassady and Whitley (425) and Johnson (426). Table 7.15 lists some of the more uncommon viral pathogens that have been associated with CNS disease.
frequency, fatality rates, and laboratory confirmation, as derived from data from Cassady and Whitley (425) and Johnson (426). Table 7.15 lists some of the more uncommon viral pathogens that have been associated with CNS disease.
TABLE 7.13 Known Reported Causes of Encephalitis in 412 Children 16 Years or Younger (1968 Through 1987) | ||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||
TABLE 7.14 Common Viral Infections of the Central Nervous System | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 7.15 Viruses That have been Rarely Associated with Central Nervous System Disease | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Enterovirus Infections
Enterovirus (a group of RNA viruses) infections include poliomyelitis (3 types), coxsackie groups A (22 serotypes) and B (6 serotypes), and the echovirus group (31 serotypes). Viruses isolated after the original classification are simply assigned an enterovirus number (serotypes 68 to 72) (427).
Organisms enter the body through the oral or respiratory tracts, replicate, and extend to the regional lymphoid tissue, induce a minor viremia, and subsequently can invade multiple sites, including the nervous system. The clinical picture produced by these agents depends on factors such as type and virulence of the virus, the site of CNS invasion, and, as is the case for poliomyelitis, host factors such as degree of susceptibility and recent inoculations or trauma (428).
Enterovirus infection is transmitted principally by direct or indirect contamination through infected fecal material. Organisms can be isolated from the oropharynx, stool, blood, or CSF. Isolation of virus from CSF confirms the cause of a specific episode (428). Diagnosis by isolation of an enterovirus from stool can be misleading because virus can be excreted for 12 to 17 weeks after infection. In such a situation, only a definite increase in the titer of antibodies to the isolated agent can confirm a recent significant infection.
The clinical manifestations of an enterovirus infection of the nervous system can take several forms.
In aseptic meningitis, children usually have an infection associated with epidemic disease caused by coxsackieviruses A9 and B2, 4, and 5 or echoviruses 4, 6, 9, 11, 16, 30, and 33; other types, such as echoviruses 18, 7, and 13, have also emerged in outbreaks (429). Febrile infants infected with coxsackievirus group B and echoviruses 11 and 30 were more likely to have aseptic meningitis than those infected with other serotypes (430). Berlin and coworkers were able to identify the viral pathogen in 62% of children younger than 2 years of age with aseptic meningitis; group B coxsackievirus or echoviruses were implicated in 92% of the laboratory-diagnosed cases (431). In 63.5% of cases, the infection developed at 8 weeks of age or earlier (432). Fever, pharyngitis, or other respiratory symptoms are common. Abdominal pain can occur. Rash is common but its incidence depends on the specific viral agent; one-third to one-half of patients with echovirus 9 meningitis have exanthem. This rash is variable in appearance, but can be petechial and simulate the rash of meningococcemia. Herpangina, pleurodynia, or myocarditis are associated findings that suggest enterovirus infection (433). SIADH occurs in approximately 9% of children with enteroviral meningitis. It usually lasts less than 2 days (428,431). Multiple attacks of enteroviral aseptic meningitis occasionally have occurred in the same individual.
Enterovirus encephalitis is most commonly caused by echovirus type 9, less commonly by echoviruses 4, 6, 7, 11, and 30 and coxsackievirus B5. Outbreaks of encephalitis have been associated with enterovirus 71 (434) where systemic inflammatory responses may trigger cardiopulmonary collapse (435), especially in younger children (436,437,438,439). Three neurologic syndromes have been observed. Most commonly, there is a rhombencephalitis marked by myoclonic jerks, ataxia, and various brainstem signs; less often there is an aseptic meningitis or an acute flaccid paralysis (434,440). Enteroviruses account for approximately 10% to 20% of the cases of encephalitis of proven viral etiology. Neurologic symptoms can be diffuse or focal (441). Clinical manifestations of the diffuse form of encephalitis range from mild changes of mental status to coma, whereas focal neurologic symptoms include focal seizures, hemichorea, or ataxia. Although the general prognosis is favorable, fatal cases of enterovirus encephalitis involving coxsackieviruses B3 and B6, echoviruses 2, 9, 17, and 25, and enterovirus 70 have been reported (428,431). There is some evidence of efficacy of the antiviral agent pleconaril in enteroviral meningitis (438,442).
Coxsackieviruses and echoviruses can on rare occasions cause paralytic syndromes that are clinically indistinguishable from paralytic poliomyelitis. The course is less severe, however, and of shorter duration. Acute paralytic syndromes also can occur after acute epidemic conjunctivitis caused by enterovirus type 70 (443). The three patterns most commonly seen are limb involvement only (43%),
involvement of nerves and limbs (33%), and involvement of one or more cranial nerves only (24%). Severe, diffuse pain that does not conform to root or nerve distribution and that is sometimes associated with muscle tenderness usually precedes the onset of paralysis (444). Approximately one-half of patients have fasciculations during the acute paralytic stage. In some instances, radiculitis, myelitis, or radiculomyelitis can occur (445).
involvement of nerves and limbs (33%), and involvement of one or more cranial nerves only (24%). Severe, diffuse pain that does not conform to root or nerve distribution and that is sometimes associated with muscle tenderness usually precedes the onset of paralysis (444). Approximately one-half of patients have fasciculations during the acute paralytic stage. In some instances, radiculitis, myelitis, or radiculomyelitis can occur (445).
Coxsackieviruses and echoviruses have been associated with a variety of other neurologic conditions, notably Guillain-Barré syndrome, transverse myelitis, dermatomyositis, polymyositis, cerebellar ataxia, and the syndrome of opsoclonus-myoclonus (431,446,447). Pulmonary edema, thought to be neurogenic in origin, has been associated with enterovirus 71 (448). Cardiopulmonary failure occurs in the absence of severe neurologic signs but is accompanied by severe, widespread inflammation of the brainstem and spinal cord (449).
Enterovirus infections of neonates are a serious and potentially fatal condition. Vertical transmission of enteroviruses can occur transplacentally, during delivery, and during the postpartum period. Whereas the presence of a primary maternal infection during late pregnancy can lead to stillbirth and severe neonatal disease manifested within the first 2 days of life, this is not a common occurrence. The majority of children manifest symptoms between the third and tenth days of life. This reflects the transmission of the virus by genital, respiratory, and gastrointestinal secretions during the immediate perinatal period. Enteroviruses reported to cause neonatal disease include coxsackieviruses B1 through B5 and echoviruses 5, 7, 9, 11, 17, 19, and 22 (450).
Signs and symptoms suggestive of severe CNS involvement include extreme lethargy, flaccid paralysis, and coma. These symptoms usually are associated with a multisystem organ involvement that includes liver, heart, and lungs with hypocoagulability and bleeding, a picture suggestive of neonatal sepsis (451). In the experience of Rorabaugh and associates, only 8.7% of infants younger than 16 weeks of age show meningeal irritation when first examined (432). A more benign picture of isolated aseptic meningitis also has been seen. Febrile seizures might be caused by enteroviral infection in the central nervous system, especially in summer months (452). Because of the decline in neonatal bacterial meningitis, enteroviruses have become the most common cause of meningitis in neonates older than 7 days and are responsible for one-third of all cases of neonatal meningitis (453). Repeat episodes of meningitis caused by different strains of enterovirus can occur within 1 month of each other (454).
A long-term follow-up study suggests that approximately 10% to 15% of infants who recover from enterovirus infections incurred during the first year of life have motor deficits or subnormal intelligence (455,456,457). Enterovirus infections in infants younger than 3 months of age can cause subtle deficits in language skills, particularly in receptive language. These children, therefore, should be monitored carefully for language development (458). When infection incurred after the first year of life, no residua are usually demonstrable.
Persistent and fatal enteroviral CNS infections can occur in patients with agammaglobulinemia (441,459).
The diagnosis of enteroviral infections is based in the recovery of the virus in feces, urine, blood, or CSF or from cultures obtained on biopsy specimen during the acute episode of the disease (460,461). The PCR technique is highly sensitive and specific for the detection of virus-specific DNA. Its speed, accuracy, and sensitivity make it superior to virus culture of CSF for the diagnosis of enterovirus meningitis (462,463).
Poliomyelitis
Although poliomyelitis caused by wild virus has been eradicated from the Western hemisphere since 1994, it remains a problem in developing countries. An extensive discussion is warranted because it serves as a prototype for viral infections of the nervous system.
Poliomyelitis is an acute infectious disease first described by Heine in 1840 (464). It affects the motor neurons of the spinal cord and brain and results in an asymmetric flaccid paralysis of the voluntary muscles. The severity of the illness varies; asymptomatic or mild cases are approximately 100 times more common than the classic paralytic disease. Poliomyelitis can occur throughout the world. It is contracted earlier in life and is less likely to be symptomatic in areas with inadequate sanitation because poor sanitation is conducive to exposure at an age when lingering transferred maternal immunity can attenuate the clinical picture.
With widespread immunization, poliomyelitis has become preventable, and recurrent major epidemics are no longer encountered. Approximately 8 to 10 cases per year have been reported in the United States. All of these have been associated with the use of the oral polio vaccine (OPV). These cases are more commonly associated with the administration of the first dose of vaccine (1 per 720,000 recipients of OPV) (465,466). Based on the lack of wild cases and the occurrence of vaccine-associated paralytic polio (VAPP), the Advisory Committee on Immunization Practices voted to recommend a sequential schedule of inactivated polio vaccine (IPV) and OPV for polio immunization, with either all OPV or all IPV as acceptable alternatives (467).
Pathology
Alterations found within the CNS depend on the stage of the disease at the time of death. In the early days of the illness, neurons undergo a variety of nonspecific microscopic
changes that vary in severity and result directly from viral invasion. These changes are accompanied by an initial polymorphonuclear reaction, which later becomes mononuclear. As the illness progresses, motor neurons degenerate, become surrounded by inflammatory cells, and undergo neuronophagia. The distribution of lesions is characteristic of poliomyelitis. Lesions are usually mild and restricted to layers 3 and 5 in the precentral gyrus and adjacent cortex, thalamus, and globus pallidus (468). Neuronal necrosis can be caused by toxic levels of neurotransmitters such as quinolinic acid that occur in areas of the brain affected by the virus as well as from direct viral damage to neurons (469).
changes that vary in severity and result directly from viral invasion. These changes are accompanied by an initial polymorphonuclear reaction, which later becomes mononuclear. As the illness progresses, motor neurons degenerate, become surrounded by inflammatory cells, and undergo neuronophagia. The distribution of lesions is characteristic of poliomyelitis. Lesions are usually mild and restricted to layers 3 and 5 in the precentral gyrus and adjacent cortex, thalamus, and globus pallidus (468). Neuronal necrosis can be caused by toxic levels of neurotransmitters such as quinolinic acid that occur in areas of the brain affected by the virus as well as from direct viral damage to neurons (469).
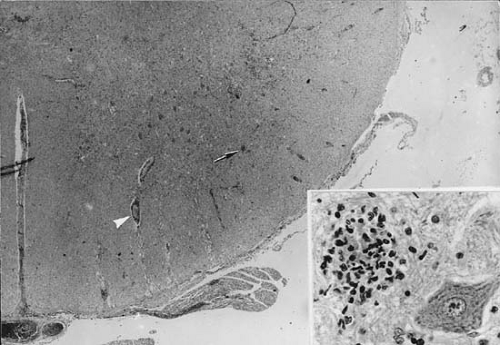 FIGURE 7.9. Poliomyelitis, acute stage. Anterolateral quadrant of the spinal cord at lumbrosacral enlargement shows widespread inflammation, mainly in the anterior horn. Perivascular cuff of chronic inflammatory cells (white arrowhead) and neuronophagia (black arrow) are apparent (H and E stain, ×22). Inset shows details of neuronophagia (hematoxylin and eosin stain, ×450). West Nile encephalitis shows a similar picture. (Courtesy of Dr. Hideo H. Itabashi, Departments of Pathology and Neurology, Los Angeles County Harbor Medical Center, Torrance, CA.) |
Cerebellar vermis and deep cerebellar nuclei are often severely involved, whereas the cerebellar hemispheres are usually free of lesions (470). In addition, marked involvement of motor and sensory cranial nuclei of the medulla occurs, especially the vestibular and ambiguous nuclei and throughout the reticular formation. The basis pontis and inferior olivary nuclei are usually spared.
In the spinal cord, lesions usually are restricted to the anterior horn cells, although some cases also show spotty involvement of the neurons in the intermediate, intermediolateral, and posterior gray columns (Fig. 7.9). Extension of lesions to the sensory spinal ganglia is the rule in more extensive cord involvement. The cervical and lumbar cords tend to be more affected than the thoracic region.
Clinical Manifestations
The clinical picture of poliomyelitis ranges from a nonspecific mild febrile illness to a severe and potentially fatal paralytic disease. An inapparent infection is estimated to occur in 90% to 95% of infected individuals, a minor nonparalytic illness in 4% to 8%, and paralytic poliomyelitis in 1% to 2% (Fig. 7.10). The incubation period ranges between 3 and 35 days, with an average of 17 days (471).
The minor illness coincides with the period of viremia and the appearance of virus in the stool. It lasts 24 to 48 hours and is manifested by nonspecific symptoms of headache, vomiting, sore throat, and gastrointestinal disturbances.
 FIGURE 7.10. Schematic diagram of the clinical and subclinical forms of poliomyelitis, showing presence of virus and antibodies in relation to the development and subsidence of infection. (From Horsfall FL, Tamm I. Viral and rickettsial infections of man, 4th ed. Philadelphia: Lippincott, 1965. With permission.) |
The major illness can follow in 2 to 5 days of recovery from the minor illness. When mild, it produces aseptic meningitis with fever, headache, vomiting, and meningeal irritation. The disease might not advance any further or it might progress to the paralytic stage.
The clinical course and picture of paralytic poliomyelitis varies with the age of the patient and the site of maximal involvement. The classic diphasic course is more common in children than in adults. It was noted in 37% of children aged 2 to 9 years but only in 11% of adolescents 15 years or older (472). In the remaining cases, the preparalytic stage is mild or inapparent. Infants show relatively little fever and lack meningeal signs despite extensive and often symmetric paralysis. Paralysis occurs in 90% of infants younger than 1 year of age who develop symptomatic infections with poliomyelitis virus (473).
The presenting symptom of paralytic poliomyelitis is pain, often severe and localized to the lower back. It usually is accompanied by hyperesthesia, drowsiness, or irritability. The initial indications of paralysis are localized muscular pain, fasciculation, twitching, and diminution or loss of the deep tendon reflexes. The paralysis appears during the first or second day of the major illness. Often it reaches its maximum within a few hours, and, less commonly, it can advance for 4 to 5 days. The fever that generally accompanies the paralysis falls by lysis.
Table 7.16 shows the distribution of paralysis, as encountered in an early epidemic (474). Curiously, when an infected child had received an injection before the onset of symptoms, the illness more likely involves the injected limb. A similar problem has been encountered when intramuscular injections are given within 30 days of immunization with oral poliovirus vaccine (475). These observations can be explained by the upregulation of the poliovirus receptor consequent to trauma. The poliovirus receptor is an integral membrane protein with structural characteristics of the immunoglobulin superfamily of proteins (476,477).
TABLE 7.16 Distribution of Paralysis in 5,784 Cases of Acute Anterior Poliomyelitis | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||
Spinal, bulbar, and encephalitic forms of poliomyelitis have been recognized. Spinal paralytic poliomyelitis is characterized by an asymmetric, scattered, flaccid paralysis that more frequently involves the lower extremities. On examination, the weakness is accompanied by muscle tenderness and spasm. Deep tendon reflexes in the affected area are usually absent. Autonomic involvement can be manifested by excessive sweating and vasomotor disturbances. Some 10% to 20% of patients have bladder involvement, notably sphincter spasm and overflow incontinence (478). Pyramidal tract and segmental sensory involvement are extremely rare.
The bulbar form of poliomyelitis generally evolves with explosive rapidity. Other areas of the neuraxis, most often the cervical cord, also are affected in 90% of cases.
The clinical picture of bulbar poliomyelitis reflects the site of involvement. Most commonly, the paralysis affects the facial, pharyngeal, and ocular muscles (see Table 7.16) (474,479). Involvement of the reticular formation can lead to respiratory and cardiovascular irregularities (480). Pulmonary edema is regarded as a manifestation of extensive damage to both the dorsal nuclei of the vagus and the medial reticular (vasomotor) nuclei of the medulla (481). When the tenth cranial nerve is involved, there is impairment of swallowing and faulty innervation of the larynx. This presents a serious threat to life by obstruction of the airway and is an indication for early elective tracheotomy. Fatal involvement of the respiratory center or the circulatory center may occur. Hypertension can develop secondary to medullary involvement or hypocapnia. Hypothalamic dysfunction is manifested by impaired temperature regulation and hypertension (482). Papilledema results from carbon dioxide retention caused by inadequate pulmonary ventilation. It also is associated with a high CSF protein, which interferes with normal CSF absorption.
In as many as one-third of cases, poliomyelitis is accompanied by symptoms and signs of encephalitis, including changes in consciousness, anxiety, irrationality, and restlessness or hyperactivity, muscular tremors, and twitching. These signs should first be considered as caused by hypoxia even in the absence of cyanosis. Only when these symptoms persist after adequate oxygenation can they be attributed to viral involvement of the cortex. Other complications of the acute stage of poliomyelitis include infections of the respiratory tract and atelectasis.
Paralytic ileus with gastric atony can result from viral damage to the vegetative centers or altered serum potassium concentrations.
Diagnosis
The blood is usually normal, although leukopenia can be found in the early stages of the illness. The CSF is almost always abnormal. CSF pressure is normal but can become elevated with carbon dioxide retention. The cell count is
highest in the preparalytic stage, when polymorphonuclear cells predominate. The average count of 185 cells/μL decreases to 50 cells/μL at 1 week after the onset of paralysis. By then there is a predominantly monocytic response. The protein content is normal in the early stages of the illness. In the experience of Merritt and Fremont-Smith, it reached a maximum 25 days after the onset of paralysis (483). Thereafter, the protein content decreased gradually, remaining elevated long after the pleocytosis had disappeared. The sugar content is normal.
highest in the preparalytic stage, when polymorphonuclear cells predominate. The average count of 185 cells/μL decreases to 50 cells/μL at 1 week after the onset of paralysis. By then there is a predominantly monocytic response. The protein content is normal in the early stages of the illness. In the experience of Merritt and Fremont-Smith, it reached a maximum 25 days after the onset of paralysis (483). Thereafter, the protein content decreased gradually, remaining elevated long after the pleocytosis had disappeared. The sugar content is normal.
The virus is best isolated from the stool and oropharynx. Poliovirus has been isolated from the stool as early as 19 days before onset of illness and as late as 3 months after onset. The mean duration of virus excretion is approximately 5 weeks after onset of clinical illness. Poliovirus is rarely isolated from the CSF. The PCR technique performed on CSF (484), stool, blood, and throat specimens (485) is a sensitive, specific, and rapid technique for the diagnosis of enteroviral infections. It also has been used to differentiate wild poliovirus infections from vaccine strains and from other enteroviral infections (486). Serologic diagnosis is impractical but can be made by demonstrating an elevation in the specific complement fixation or neutralizing antibody titers. The etiologic diagnosis of inapparent, abortive, or nonparalytic poliomyelitis can be made only by laboratory studies.
Currently, poliomyelitis occurs rarely, and no confirmed case of wild-type polio virus infection has been reported in the United States since 1991 (487). Since then, all cases encountered in the United States have been associated with vaccination (488,489). All patients who develop poliomyelitis soon after oral vaccine administration should be evaluated for humoral and cellular immunosuppression (490). In the survey of Nkowane and coworkers, 33% of cases of poliomyelitis encountered between 1973 and 1984 were seen in recipients of OPV, 48% in contacts of such recipients, and 13% in immunodeficient patients (488). Virus reversion to the neurovirulent form involves point mutations at a single or several nucleotide positions (491).
Differential Diagnosis
In patients who are not immunodeficient and who have not been exposed to the vaccine, the diagnosis of poliomyelitis has become difficult. A number of other enteroviruses, notably coxsackieviruses and echoviruses, can produce a flaccid paralysis. When these conditions are unassociated with an exanthem, their diagnosis rests on PCR, viral isolation, or serologic confirmation. An illness resembling acute poliomyelitis also has been observed after infection with Epstein-Barr virus (494) as well as with an agent of the Russian spring-summer louping ill group of viruses (495).
Poliomyelitis must be differentiated from infectious polyneuritis, which has as presenting symptom a symmetric paralysis, usually progresses for more than 3 days, and is unaccompanied by fever. When sensory deficits can be documented, they confirm the diagnosis of polyneuritis. The absence of pleocytosis in the presence of a progressive paralysis also argues for polyneuritis.
A poliomyelitis-like paralysis (Hopkins syndrome) can occur during recovery from an asthma-like respiratory tract disease (496) (see Chapter 17). In some instances this syndrome can result from enterovirus or herpesvirus type 1 infections (497). Other conditions that mimic poliomyelitis include epidemic motor polyradiculoneuritis, insect and snake bites, schistosomiasis of the spinal cord, ingestion of chemicals such as arsenic and organic phosphates, trichinosis, and tetanus (498,499,500).
Prognosis
Recovery from nonparalytic forms of poliomyelitis is complete. The degree of disability that results from paralytic poliomyelitis depends on the extent of involvement. Function generally improves as pain and spasm recede after the acute phase. Overall mortality for the paralytic disease is approximately 4%. Spontaneous recovery of muscle function begins within a few weeks. Some involved neurons recover, but muscles totally paralyzed 1 month after the onset of the illness become completely normal only 1.4% of the time (501). Muscles severely paralyzed at the end of the acute period regain functional strength in 50% of cases and become normal in 20%. Moderately paralyzed muscles recover in 90% of cases. One-half the affected muscles continue to improve after 1 year. Three-fourths of functional recoveries occur in the first year, slightly less than one-fourth during the second year, and 5% during the third year. In general, prognosis is best for the proximal muscles in the lower extremities; it is poorest for the opponens pollicis and abdominal muscles (502).
Children with severe paralytic disease have developed a progressive neurologic disorder marked by increased weakness, fatigability, and pain some 30 to 40 years after the initial infection. Several explanations for this postpolio syndrome have been proposed. Sharief and colleagues suggested that it results from a chronic progression of the infection because there is intrathecal production of detectable IgM oligoclonal bands specific for poliovirus in the CSF and elevated CSF levels of IL-2 (503). Mononuclear perivascular cuffs occur in postpolio patients dying as long as 38 years after poliomyelitis (504). Late decompensation also could result from the combination of an abnormal enlargement of the motor units, resulting from the loss of the anterior horn cells, and the increased end-plate complexity seen with aging (504,505). Another
factor that could cause postpolio syndrome is chronic muscle overuse because 40% of children with severe residual paralytic disease have elevated creatine kinase levels (505). Patients with residual leg weakness are twice as likely to report symptoms in those limbs as those with residual arm weakness (506). Sleep apnea can be a late complication of the postpolio syndrome (507). Relapses and second attacks of poliomyelitis are extremely rare. Other than a continued exercise program, there is no effective treatment for this condition (508).
factor that could cause postpolio syndrome is chronic muscle overuse because 40% of children with severe residual paralytic disease have elevated creatine kinase levels (505). Patients with residual leg weakness are twice as likely to report symptoms in those limbs as those with residual arm weakness (506). Sleep apnea can be a late complication of the postpolio syndrome (507). Relapses and second attacks of poliomyelitis are extremely rare. Other than a continued exercise program, there is no effective treatment for this condition (508).
Treatment
Control of poliomyelitis has been achieved by the use of orally administered live attenuated virus vaccine. Because of the vaccine-associated paralytic polio, changes in the poliovirus vaccine schedule occurred in 1997. Details of the advised vaccination schedules are presented in the 1997 Report of the Committee on Infectious Diseases, American Academy of Pediatrics (509).
Oral attenuated poliomyelitis vaccine should not be given to individuals with altered immunocompetence. Inactivated poliomyelitis vaccine is recommended for adults 18 years or older because of the increased risk of vaccine paralysis in that age group (488,489) and for immunization of HIV-positive children (509). For a detailed discussion of the response to vaccination, the characteristics of immunity, and the complications of vaccination, the reader is referred to Salk and Salk (510).
During the acute phase of the illness, treatment is solely symptomatic.
Coxsackievirus
An increasing number of coxsackievirus strains have been reported as causing infections of the human nervous system. The clinical pictures produced by the various strains of coxsackievirus are indistinguishable. Neurologic manifestations include aseptic meningitis, encephalitis, acute flaccid paralysis, Guillain-Barré syndrome, transverse myelitis, cerebellar ataxia, opsoclonus-myoclonus, and epidemic pleurodynia (511). These occur with both group A and B infections.
The clinical picture of aseptic meningitis caused by infection with coxsackievirus is nonspecific. The illness is preceded by a prodromal phase manifested by malaise and gastrointestinal disturbances, during which time viremia can be demonstrated. Neurologic symptoms can develop in association with pleurodynia, particularly in older children infected with B1 and B3 viruses. Exanthems are rare.
Coxsackieviruses B5, 1, 2, and 4 and A9 have been associated with the development of a clinical picture of encephalitis. The outcome is usually favorable. Serotypes A5, A9, and B2 have been associated with focal encephalitis (512).
Group A viruses also can cause acute lymphonodular pharyngitis, acute parotitis, hand-foot-and-mouth disease, polymyositis, chronic myopathies, and respiratory infections. Group B viruses have been associated with pleurodynia (Bornholm disease or epidemic pleurodynia) and diarrhea. Both types have been associated with pericarditis and myocarditis.
Anatomic alterations within the CNS are slight and nonspecific. They include meningeal inflammation and focal glial infiltration of gray and white matter of cerebrum, brainstem, and cerebellum (513).
A mild paralytic disease has been reported (514,515) as well as acute cerebellar ataxia, unilateral paralysis of the oculomotor nerve, palatal paralysis, and a postencephalitic parkinsonian picture (515,516). Focal necrotizing encephaloclastic lesions with subsequent porencephalic cyst formation also can occur.
Coxsackievirus infection is diagnosed by recovering the virus from blood or CSF, feces, and oropharyngeal swabbings, by detection of viral DNA by PCR in CSF or in brain biopsies, and by demonstrating an at least fourfold increase in neutralizing antibodies against the isolated virus.
The prognosis for recovery is usually excellent, and treatment is purely symptomatic.
Echovirus
Echoviruses are the most frequent cause of enterovirus infection of the nervous system. Sporadic infections have been described for 25 members of the group, whereas outbreaks usually occur with types 4, 6, 9, 11, 16, 18, and 30.
The clinical manifestations vary. Most common is an aseptic meningitis. All echovirus serotypes except 24, 26, 29, and 32 are associated with this entity (517). The illness is biphasic and generally self-limited. It presents a nonspecific picture of fever, headache, signs of meningeal irritation, sore throat, neck and back pain, and, occasionally, myalgia. In some cases, symptoms simulate basilar migraine (518). The illness usually resolves in 7 to 10 days. Certain viral strains can produce an illness associated with exanthematous or petechial rashes that can antecede or be associated with the period of central nervous involvement.
Although most patients have no paralytic residua, a few patients have been left with impaired strength in the anterior neck muscles. Rarely, a paralytic disease indistinguishable from poliomyelitis can occur with echovirus infections. Paralysis can be mild and transient or so severe as to be permanent or fatal. In the latter case, the virus has been isolated from CNS tissue.
Encephalitis manifested by focal or generalized seizures, disorders of sensorium, tremor, cranial nerve deficits, disorders of ocular movement, choreiform movements, paresthesias, and other signs of cerebral dysfunction have been associated with echovirus infections (519). Cerebellar ataxia of short duration (520) and acute
ascending polyradiculomyelitis also have been described (521) as has muscular hypotonia in association with bilateral edema of the basal ganglia (522). Encephalitis with white matter necrosis can occur in the neonatal period (523). Persistent and fatal CNS infections with echoviruses can occur in patients with agammaglobulinemia (524). The course of these infections can be ameliorated by repeated administration of intravenous infusions of gamma globulin and intrathecal administration of gamma globulin prepared for intravenous use (524,525).
ascending polyradiculomyelitis also have been described (521) as has muscular hypotonia in association with bilateral edema of the basal ganglia (522). Encephalitis with white matter necrosis can occur in the neonatal period (523). Persistent and fatal CNS infections with echoviruses can occur in patients with agammaglobulinemia (524). The course of these infections can be ameliorated by repeated administration of intravenous infusions of gamma globulin and intrathecal administration of gamma globulin prepared for intravenous use (524,525).
The diagnosis of echovirus infection usually rests on laboratory tests. IgG echovirus antibody elevation has been demonstrated in CSF of individuals with and without meningeal involvement. Because antibody titers to other viruses also are elevated, IgG echovirus antibody elevation reflects a nonspecific transport of IgG-associated viral antibodies from the serum to the CSF or reflects endogenous production of multiple antibodies within the CNS (526). PCR of CSF and a fourfold rise of specific antibodies confirm the etiologic agent.
Newer Enteroviruses
Four new enteroviruses (serotypes 68 through 71) have been recognized. These viruses can cause sporadic or epidemic infections of hand-foot-and-mouth disease and they have been recognized throughout the world. Serotypes 70 and 71 have been associated with CNS infections such as aseptic meningitis, encephalitis, and encephalomyelitis. Serotype 71 is responsible for more serious CNS disease, leading to acute paralytic manifestations indistinguishable from poliomyelitis and fatal encephalomyelitis (433,434,527).
The diagnosis is based on the virus isolation from skin vesicles, CSF, and brain tissue, using a monoclonal fluorescent antibody technique. No specific serologic diagnosis exists. Treatment of these infections is supportive.
Adenoviruses
Adenovirus infections usually cause an acute respiratory disease associated with conjunctivitis or keratoconjunctivitis. In a small percentage of cases, these symptoms are associated with involvement of the nervous system, notably a severe encephalitis or meningoencephalitis (529,530). A syndrome of reversible encephalopathy has also been reported (531). Neonatal infections usually are generalized. In fatal cases with neonatal encephalitis the CSF has been reported to be normal (532).
The diagnosis of these infections can be made through the isolation of the virus from the CSF or brain. A fourfold rise in adenoviral antibodies can provide a retrospective diagnosis. The prognosis of the CNS manifestations of adenoviruses is difficult to ascertain because of the few reported cases. No specific treatment for these infections is available.
Human Herpesvirus Types 6 and 7
Exanthem Subitum (Roseola Infantum)
Roseola, the most common exanthem of infancy, is characterized by the relative absence of prodromal symptoms, a rapid increase in body temperature, and conspicuously little listlessness or irritability. The temperature remains elevated for 3 to 5 days and subsides with the appearance of maculopapular rash. A bulging fontanel, the result of increased intracranial pressure, occurs during the second to fifth day of fever in about one-third of children (533). The condition occurs at a median age of 9 months when caused by human herpesvirus-6 (HHV-6), and at a median age of 26 months when caused by human herpesvirus-7 (HHV-7) (534). Other viral agents also are believed to be responsible for this condition (535).
CNS involvement can occur from 1 day before the onset of fever to day 2 of the illness (533). Neurologic complications include aseptic meningitis, acute disseminated encephalomyelitis (536,537), acute encephalitis (538), and acute cerebellitis.
The incidence of seizures varies considerably from one series of cases to the next (539). In the prospective study of Hall and coworkers, HHV-6 infection accounted for one-third of all febrile seizures younger than the age of 2 years (540). Persistent neurologic complications and death have been reported. The former include hemiparesis, recurrent seizures, and mental retardation. The cause of the residual encephalopathy is unknown (536).
HHV-6 DNA has been detected in the CSF of children who had three or more febrile seizures. This observation suggests that in some instances recurrence of febrile convulsions is associated with reactivation of the virus (540,541,542).
Approximately one-fifth of children have CSF with either a pleocytosis of more than 5 white blood cells/μL or a protein of more than 45 mg/dL; CSF glucose is normal. Approximately one-fourth of children have encephalopathy or encephalitis; all of these have an abnormal EEG. Transient periodic complexes can occur over the temporal lobes similar to those described for herpes simplex virus encephalitis (536). Unilateral or bilateral CT changes usually begin as hypodense areas that can progress to irregular cerebral atrophy or become hemorrhagic (536).
The association of HHV-6 with the neurologic complications is suggested by the concomitant isolation of virus, the identification of DNA by PCR in the CSF (543), and the fourfold increase in specific neutralizing antibodies. HHV-6 reactivations include a number of neurologic complications later in life (544).
HHV-7 has been associated with febrile seizures, a severe encephalopathy, and in two instances acute hemiplegia by the isolation of HHV-7 through viral culture and PCR (545,546). The diagnosis of HHV-7 is based on the methods described for HHV-6 (547).
Many strains of HHV-6 are sensitive in vitro to ganciclovir and foscarnet (548). Doses of 10 mg/kg per day in two divided doses of ganciclovir or 180 mg/kg per day in three divided doses of foscarnet can be given in cases of serious CNS infections caused by HHV-6.
Arboviruses
Arthropod-borne (arbo) viruses are RNA viral agents transmitted between susceptible vertebrate hosts by means of blood-sucking arthropods (549). Approximately 200 viruses have been tentatively classified into this group, and approximately 150 of these have been assigned to 21 groups that share common antigens. At least 51 have been associated with human disease. The epidemiology of these infections is largely determined by the nature and lifestyle of the vector. Most of the infections are sustained in sylvan hosts, with transmission through humans and domestic animals being usually incidental and insignificant in the natural history of the virus. Of greatest significance in the United States are the La Crosse (LAC) strain of the California encephalitis group, St. Louis encephalitis (SLE), western equine encephalitis (WEE), eastern equine encephalitis (EEE), Venezuelan equine encephalitis (VEE), Colorado tick fever, West Nile virus, and Powassan encephalitis. In this section we also include Dengue virus and Japanese encephalitis virus.
In humans, these viruses can be asymptomatic or can produce a mild generalized illness. The full-blown diseases, which are discussed in this section, are the least common manifestations of infection.
The nonspecificity of symptoms necessitates diagnosis by means of laboratory studies. Viral-specific IgM antibodies appear in serum in the first week of illness, and serologic tests on paired samples taken several weeks apart can suggest a recent infection (550). The detection of virus-specific IgM in the CSF is diagnostic. Virus can be isolated from blood or from nervous tissue by means of biopsies or autopsies.
La Crosse Encephalitis
LAC, a member of the California serogroup of Bunyavirus, is the most commonly reported cause of mosquito-borne disease in the United States (551). This disease, originally encountered most frequently in Midwestern states, also has become endemic in the mid-Atlantic region. Since 1993, West Virginia has had more reported cases that any other state (552). Aedes triseriatus, the principal mosquito vector, transmits the virus during the summer and early fall. Tree holes that contain water as well as discarded automobile tires that hold rainwater serve as the breeding habitat for the mosquito. Unlike St. Louis encephalitis virus and other arboviruses that cause epidemic disease, La Crosse virus infections exhibit an endemic pattern.
In 99% of instances, LAC virus infection is asymptomatic. The symptomatic disease can be mild or severe (553). LAC is the most prevalent and most pathogenic of the California serogroup of viruses (554,555).
Pathology
The alterations within the brain are largely microscopic. They are those of an acute encephalitis without distinctive findings (553).
Clinical Manifestations
The peak incidence of LAC is between July and October. There is a strong association between rural outdoor exposure and the presence of antibodies and clinical illness; the incidence of antibodies is 26.8% in rural populations and 15.3% in urban areas. Significant increases in antibody titer occur between ages 5 and 9 years among rural populations and between ages 10 and 19 years among urban populations living in endemic areas (556). Male individuals are affected three times as often as female individuals.
The clinical picture is that of a meningoencephalitis with fever, changes in consciousness, headache, and meningeal irritation (553,557,558). Balfour and associates defined two clinical patterns (559). A mild form begins with a 2- to 3-day prodromal stage characterized by fever, headache, malaise, and gastrointestinal symptoms. An increase in temperature on approximately the third day is associated with lethargy and evidence of meningeal irritation. Symptoms abate over 7 or 8 days without overt neurologic residua.
The severe form begins abruptly with fever and headache, followed in 12 to 24 hours by sudden onset of focal or generalized seizures. These have been reported in 40% to 50% of patients with neurologic symptoms (559). Hypernatremia and increased body temperature may be related to clinical deterioration (560).
The CSF shows pleocytosis, which in 85% of cases is mainly mononuclear. The protein content is mildly elevated in 20% of the patients and the glucose content is normal. The LAC PCR test for cerebrospinal fluid enables rapid diagnosis (554). Presence of the SIADH is common. The EEG is always abnormal, with abnormalities persisting for months or years in 55% of cases (561).
Diagnosis
In contrast to infections caused by enteroviruses, LAC is not biphasic and is not obviously associated with upper respiratory signs or symptoms. Familial outbreaks are extremely rare. EEG and neuroimaging demonstrate a focal process in 15% to 40% of patients (559). Such focal involvement also is observed in herpes simplex encephalitis; in both conditions, the CSF can be hemorrhagic. Specific
IgM and IgG serologies in CSF and serum confirm the viral etiology. A fourfold increase in these antibodies provides a retrospective diagnosis. If a brain biopsy is performed, infected brain cells can be detected by a monoclonal antibody technique (562).
IgM and IgG serologies in CSF and serum confirm the viral etiology. A fourfold increase in these antibodies provides a retrospective diagnosis. If a brain biopsy is performed, infected brain cells can be detected by a monoclonal antibody technique (562).
Prognosis
LAC is a relatively mild illness, with a case fatality rate in symptomatic cases of less than 1% (551). Permanent neurologic residua are uncommon, but patients can have a variety of neurologic and behavioral residua (558,563). Older children and adolescents tend to show emotional lability, difficulty in learning, and personality problems. Later, difficulties in processing visual and auditory information become apparent, and some children develop behavior that resembles the organic hyperkinetic syndrome (564). However, a controlled assessment of 29 children who recovered from encephalitis failed to demonstrate significant personality changes when compared with the premorbid state (564).
Treatment
The treatment of neurologic manifestations of LAC has changed over the last few years. Ribavirin, which is active in vitro against LAC (565,566), was given for 10 days to a child for a severe, biopsy-proven case of LAC encephalitis, with a good outcome (565). Future studies will determine the need for its routine use.
St. Louis Encephalitis
SLE was first recognized in St. Louis in the summer of 1932 during an epidemic of encephalitis that originated in Paris, Illinois. Since then, sporadic outbreaks have occurred in the St. Louis area, California, the Rio Grande valley, Florida, and the central river valleys and western coastal regions of the United States. The vector for this virus is the Culex mosquito, and the virus has been isolated from overwintering mosquitoes (567,568).
Pathology
Diffuse vascular congestion and edema occur with meningeal and perivascular infiltration, petechiae, and glial nodules. Lesions predominate in the thalamus and substantia nigra. The cortex, anterior horns of the spinal cord, molecular and Purkinje cell layers, and dentate nucleus of the cerebellum also are involved (569).
Clinical Manifestations
Most commonly, this virus produces a clinically inapparent infection, and the ratio of asymptomatic to symptomatic cases has ranged from 19:1 to 427:1 in various outbreaks (570). Most cases occur in late summer and early fall; boys are affected three times more often than girls in some epidemics. Generally, the severity of the encephalitis and the extent of residua are greater in the older age groups (571).
The clinical manifestations of SLE range from a nonspecific flulike illness to fatal cases of encephalitis. In the 1964 Houston epidemic, most children had fever, headache, and meningeal irritation. Ataxia was noted in 27%, and fine tremors of the upper extremity in 19%. Cranial nerve palsies were encountered in 12% (570). Occasionally, the virus is responsible for transient parkinsonism (572), the syndrome of opsoclonus and myoclonus (573), and acute cerebellar ataxia (574). The latter two conditions are covered in Chapter 8.
Abnormalities in the CSF are those expected for a viral encephalitis, namely pleocytosis, which initially is predominantly polymorphonuclear, but later becomes mononuclear; a mild elevation of protein levels; and a normal sugar content (575). The EEG is often abnormal. Electromyographic studies have shown fasciculations in 59% of cases, fibrillations in 8%, and excessive insertional potentials in 54%. In some cases, these abnormalities have persisted for more than 1 month (570). The diagnosis is confirmed by specific IgM serology in the CSF or blood. When obtained within 4 days of the clinical onset of the disease, the serology has been positive in 75% of cases. Fourfold increases in specific IgG antibodies in blood can provide a retrospective diagnosis. Viral isolation should be pursued when a brain biopsy or autopsy is obtained.
Prognosis
SLE is intermediate in severity between California encephalitis and the equine encephalitides. The mortality in the 1964 St. Louis epidemic was 8% in children younger than 10 years of age. In more recent epidemics, it has ranged from 0% to 22%. Major sequelae occur less frequently than after EEE or WEE, but have been observed in 7.7% to 10.0% of survivors. Most commonly, they involve disturbances of gait and equilibrium, difficulties in speech and vision, and changes in personality (575,576). Children and young adults are much less prone to long-term motor disability than older adults (575).
Treatment
Treatment is symptomatic and supportive. No specific antiviral therapy or vaccines are available.
Western Equine Encephalitis
WEE is caused by a group A arbovirus from the family Togaviridae. It occurs in the western United States and in the central states west of the Mississippi Valley. The virus was first isolated in 1930 in California from horses with encephalitis. The chief vectors are several Culex mosquitoes, notably Culex tarsalis in the United States. The enzoonotic cycle is kept between mosquitoes, birds, and other vertebrate hosts. Horses and humans become dead-end hosts.
Pathology
The alterations within the nervous system in WEE are similar to those seen in the other encephalitides caused by arboviruses. The greatest damage is usually in the basal ganglia and cerebral white matter, with foci of demyelination and necrosis. The subcortical white matter is more extensively damaged than the cortex in the majority of fatal cases (577). White matter subjacent to cortex is more involved than deeper white matter in some children. Striking involvement of the substantia nigra might explain the relatively frequent appearance of parkinsonism as a late sequel of WEE (578,579). Extensive cystic degeneration of white matter can occur in infants.
A striking finding, noted as late as 5 years after onset of the acute illness, is the presence of both chronic inactive areas of destruction and active foci of glial proliferation and perivascular cuffing. This finding suggests that a slowly progressive form of the disease can continue for years (580).
Clinical Manifestations
Clinically inapparent infections far outnumber overt infections, although infants younger than 1 year of age have an equal incidence of apparent and inapparent infections (582).
In infants younger than 1 year of age, one common clinical picture is that of a sudden onset of high fever, seen in 92% of patients, accompanied by focal or generalized seizures, seen in 77% of patients (583). Older children have a severe encephalitis with fever, headache, vomiting, changes in consciousness, and meningeal signs. Convulsions develop in approximately 50% of children aged 1 to 4 years and in 9% of children aged 5 to 14 years.
Symptoms usually persist for approximately 10 days, then gradually subside. In fatal cases, the course is rapidly progressive. The CSF findings resemble those encountered in the other arbovirus infections (583).
The mortality in different epidemics has ranged from 9% to 23%. Sequelae are frequent and are most severe in very young infants; 56% of infants who contracted the illness when they were younger than 1 month of age had major sequelae (576). These include microcephaly, pyramidal or extrapyramidal motor impairment, seizures, and developmental retardation (580). Follow-up studies conducted on survivors reveal that the younger the patient is at the time of illness, the greater are the incidence and severity of sequelae (580,581). In older infants and in children, the sequelae are less severe. In Finley’s series, no patient contracting the illness between ages 1 and 2 years developed motor or behavior problems; 25%, however, developed seizures (580). Such seizures represent persistence of convulsions that first appeared during the acute illness.
The diagnosis is based on the detection of specific IgM and IgG in CSF and blood. The virus can be isolated rarely from blood or CSF. When brain tissue is obtained, the virus can be easily cultured.
No vaccine or specific treatment is available.
Eastern Equine Encephalitis
EEE is a group A arbovirus from the family Togaviridae first isolated in 1933 from brain tissue of infected horses. It is distributed along the Atlantic coast from the northeastern United States to Argentina. Small epidemics have been encountered in Massachusetts, New Jersey, Florida, Texas, and, more recently, Jamaica. The virus is transmitted by a variety of mosquitoes, Culiseta melanura being the most common in the United States. Birds are the amplifying host and horses and humans are dead-end hosts (550).
Pathology
The outstanding feature of EEE is the predominance of neutrophilic leukocytes in the infiltrates, which undoubtedly reflects the rapid demise of most fatally infected patients. Focal or diffuse accumulations of neutrophils and histiocytes are prominent in the leptomeninges and in the cerebral cortex, particularly the occipital and frontal lobes and hippocampus (584).
Foci of tissue damage with rarefaction necrosis permeated by neutrophilic leukocytes and pleomorphic ameboid cells are often found. Neuronolysis, often with adjacent neutrophilic leukocytes and microglial cells, is common in fulminant cases. Large perivascular collections of neutrophils, histiocytes, and other ameboid cells are detected in the white matter in regions of cortical involvement. Other sites of predilection are the basal ganglia and brainstem. Edema and congestion are prevalent early. Vascular lesions characterized by numerous small thrombi in arterioles and venules can occur. Many vessels show complete involvement of their walls with neutrophilic infiltration and fibrin deposition. As the disease progresses, the dominant cell type changes to mononuclear lymphocytes and macrophages (576,584).
Clinical Manifestations
Infected children have a subclinical to clinical ratio of 2:1 to 8:1; adults have a ratio of 4:1 to 50:1 (585). For this reason, EEE is particularly common in infants, with two-thirds of the clinical cases being younger than 2 years old. It is usually fulminant with an abrupt onset, high fever, stupor or coma, convulsions, and signs of meningeal irritation. Patients can die within 48 hours of their initial symptoms or can survive for a few days, dying from damage of the vital medullary centers, dural sinus
thrombosis, or subarachnoid hemorrhage. Survivors can remain comatose for several days or weeks before becoming responsive.
thrombosis, or subarachnoid hemorrhage. Survivors can remain comatose for several days or weeks before becoming responsive.
Laboratory studies are not diagnostic. The peripheral blood can show a prominent leukocytosis. The increase in CSF pressure can be striking. Initial CSF cell counts range between 250 and 2,000 white blood cells/μL, often containing almost 100% polymorphonuclear cells. The pleocytosis diminishes rapidly, and mononuclear cells predominate after 3 days in surviving patients (584). The EEG almost invariably shows generalized slowing and disorganization of the background activities, with the occasional patient demonstrating epileptiform discharges. The confirmation of the diagnosis is obtained through specific serology in CSF (IgM), a fourfold increase in the titer of serum antibodies, and/or the isolation of EEE virus from CSF or brain tissue (586).
Neuroimaging study results can be norma or show diffuse edema or a variety of focal lesions. In the review by Deresiewicz and coworkers, abnormal findings included focal lesions in the basal ganglia, thalami, and brainstem. Cortical lesions, meningeal enhancement, and periventricular white matter changes were less common. The presence of large radiographic lesions did not predict a poor outcome (586).
Prognosis
Infection with the virus of EEE has the most serious prognosis of all virus infections prevalent in the United States. The mortality in EEE in patients younger than 10 years is approximately 60% (585). The case fatality rate for all patients reported to the Centers for Disease Control and Prevention between 1988 and 1994 was 36%, with 35% of surviving patients showing moderate to severe sequelae. These include mental retardation, motor dysfunction, deafness, and seizures. Complete neurologic recovery is seen in only a small proportion of survivors. A high CSF white count or severe hyponatremia was predictive of a poor outcome (586).
Venezuelan Equine Encephalomyelitis
Although VEE, a group A arbovirus, is indigenous to Colombia, Venezuela, and Panama, it was responsible for the 1971 Texas encephalitis epidemic. The organism was also responsible for an outbreak of encephalitis in 1995 in Colombia and Venezuela that involved 75,000 to 100,000 cases (587). The organism has been isolated from many wild rodents as well as from Aedes and Culex mosquitoes (588).
VEE is much more benign than California (La Crosse) encephalitis, SLE, EEE, and WEE. It is marked by sudden onset of malaise, chills and fever, nausea and vomiting, headache, myalgia, and bone pain. The fever lasts from 24 to 96 hours, then abruptly drops. A period of 2 to 3 weeks of marked asthenia can follow. In contrast to other arboviruses, whose ratios of inapparent to apparent infections range from 23:1 to 500:1, in VEE a ratio of 11:1 has been reported.
The CSF findings are as in the other arbovirus infections. Virus can be isolated from serum during the first 3 days of the illness in 75% of confirmed cases (588).
PCR provides a fast, sensitive, and specific method for the detection of viral DNA in infected blood. The detection of specific IgM in CSF and serum and the observation of a fourfold increase in specific IgG in serum provide serologic confirmation of the disease.
Prognosis is good, and, with good care, fatalities are rare.
Japanese Encephalitis
Japanese encephalitis virus is transmitted by culicine mosquitoes between birds and humans. Histologic changes are most marked in the thalamus and brainstem. Patients who die rapidly can have no histologic signs of inflammation, yet have immunohistochemical evidence of viral antigen in morphologically normal neurons. Clinical features of this condition are much like those of the other encephalitides (589,590). Fever, headache, and vomiting are followed by meningism and coma. Convulsions have been reported in 85% of children. A characteristic masklike facies with a vacant stare or grimacing or lip-smacking can occur, as can a paralytic disease that mimics acute poliomyelitis. Pyramidal tract signs, a rare complication in poliomyelitis, should suggest the diagnosis of Japanese encephalitis in endemic areas (591). In the 1986 Nepal epidemic, children accounted for the majority of hospitalized cases. The fatality rate in children was markedly less than that in adults (592). In the Indian epidemic studied by Kumar and colleagues, of children with proven Japanese encephalitis, the case fatality rate was 32%; major sequelae occurred in 45%; only 29% were healthy on follow-up (593). Sequelae of the disease were more severe when the initial illness was prolonged or was associated with focal neurologic deficits. Children are more likely to have dystonia as a sequela when compared to adults (594). A TNF concentration of greater than 50 pg/mL in serum correlated significantly with a fatal outcome, whereas high levels of Japanese encephalitis virus–IgM antibodies (more than 500 units) in the CSF were associated with a nonfatal outcome (595). Elevated levels of proinflammatory cytokines and chemokines are associated with a poor outcome (596).
The MRI in Japanese encephalitis patients shows lesions in the thalamus, cortex, midbrain, cerebellum, and spinal cord. Classically, the lesions are hyperintense in T2-weighted images and hypodense in T1-weighted images (597). In some 70% of cases these are hemorrhagic. According to Kumar and coworkers, bilateral thalamic
involvement, especially hemorrhagic, can be considered to be characteristic of Japanese encephalitis in endemic areas (598). Diffusion-weighted MR imaging best demonstrates bilateral thalamic disease in Japanese encephalitis (599).
involvement, especially hemorrhagic, can be considered to be characteristic of Japanese encephalitis in endemic areas (598). Diffusion-weighted MR imaging best demonstrates bilateral thalamic disease in Japanese encephalitis (599).
A specific diagnosis can be made by detecting a fourfold increase in specific IgG antibodies in acute and convalescent sera and by measuring Japanese encephalitis virus IgM in serum and CSF by ELISA. IgM can be detected 1 week after the onset of clinical symptoms. Virus isolation can be performed but needs to be done during the first 7 days of the disease prior to the mounting of an immune response.
No effective treatment exists, and high-dose dexamethasone treatment has been of no benefit. Interferon-alpha has been used in a small number of patients, and bigger trials are being awaited to assess its efficacy. The use of sedation has been shown to independently improve outcome (600). An inverse association between interferon-gamma levels and the severity of postencephalitic sequelae has been reported (601). Motor evoked potentials may be useful in predicting motor outcome (602). A Japanese encephalitis vaccine is available in the United States (550).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


