Inherited Metabolic Disorders
Douglas R. Nordli Jr.
Darryl C. De Vivo
Introduction
There are a wide variety of disorders of metabolism, approximately 50 of which are associated with seizures or epilepsy. From the epileptologist’s perspective, many present similarly, with indistinguishable seizure semiology and electroencephalographic (EEG) findings. The clinical presentation may be influenced more by the age of the child than the specific etiology. In some children with inborn errors of metabolism and recurrent seizures, the epilepsy can be categorized into one of the recognized epileptogenic encephalopathies, such as early myoclonic epilepsy or West syndrome. In other cases, precise syndromes are lacking and terms like “generalized symptomatic epilepsy, not otherwise specified” and “epilepsies with focal and generalized features” are used. Clearly, more work needs to be done to accurately identify children with inborn errors, and we suspect that there are novel epilepsy syndromes yet to be described in this interesting group of patients. In the meantime, useful clues sometimes present that might alert one to the correct diagnosis. In some cases, this might be a relatively unique clinical feature, in others a characteristic of the epilepsy, and, finally, in some, a particular detail of the interictal EEG (Table 1). We have tried wherever possible to include these details, but we recognize that these definitive features seldom present themselves. For this reason, it is usually necessary to consider a relatively broad differential diagnosis. To organize the thought process, it is useful to think in terms of the age of the child and broad characteristics of the presentation. These have been organized into a table for ready reference (Table 2). The differential can be refined with the use of judiciously chosen screening tests. The results of these tests further guide the selection of definitive enzyme studies or genetic testing.
The acquisition of genetic information is proceeding at an exciting pace. Although this is stimulating to the neurologist, it presents a challenge for the traditional textbook format to contain up-to-date information. For this reason, we include a reference number from the Online Mendelian Inheritance of Man (OMIM) Web site so that the interested reader can quickly obtain the most current data (this source also links to Medline references). Wherever applicable, the OMIM number is given in parentheses in each heading. Another useful site is http://www.genetests.org, which provides a comprehensive listing of gene tests and the associated laboratories.
Metabolic Disorders of The Neonatal Period
The neonatal period comprises the first month of postnatal life, and a number of metabolic disorders that appear at this time include seizures as a major clinical feature.
Nonketotic Hyperglycinemia (#605899)
In this condition, glycine accumulates in the central nervous system and elsewhere in the body because of a primary biochemical defect involving the glycine cleavage system. There are four enzymes that cleave glycine: (a) P protein, a pyridoxal phosphate–dependent glycine decarboxylase (GLDC), (b) T protein, a tetrahydrofolate-requiring enzyme (GCST), (c) H protein, a lipoic acid–containing protein (GCSH), and (d) L protein, a lipoamide dehydrogenase. Disease can be caused by defects in any of these enzymes, although mutations involving GLDC and GCST are most common. Mutations that involve the pore region of the T protein appear to particularly impair function.7 The mode of transmission is autosomal recessive, with an estimated prevalence of 1/250,000. Neonatal nonketotic hyperglycinemia (NKH) is characterized by an initial variable symptom-free interval of 1 to 42 days. Clinical symptoms are at first lethargy, poor feeding, apneic spells, altered muscular tone, and intermittent ophthalmoparesis that occasionally progresses to bilateral external ophthalmoplegia. Erratic myoclonus is another major symptom that may appear early; hiccups and coma follow. The electroencephalogram (EEG) demonstrates a suppression-burst pattern that correlates with brainstem-evoked potential abnormalities and intrinsic brainstem pathology seen on postmortem examination (Fig. 1).54 Similar EEG abnormalities are characteristic of phenylketonuria, maple syrup urine disease, and molybdenum cofactor deficiency (Table 2). Clinical seizure patterns include infantile spasms and generalized tonic seizures. Progressive brain atrophy, delayed myelination, incomplete development of the corpus callosum, gyral malformations, cerebellar hypoplasia, and culpocephaly are common. Elevated glycine concentrations in the cerebrospinal fluid (CSF) during the first hours of life and a partial suppression-burst EEG pattern detected as early as 30 minutes after delivery in an asymptomatic patient indicate that the abnormal glycine metabolism has been present prenatally. A CSF-to-plasma glycine ratio >0.08 is diagnostic of nonketotic hyperglycinemia. The disease usually has a severe outcome, but milder cases have been reported. Some of these are due to abnormal splicing with slightly preserved enzyme function.72 Transient nonketotic hyperglycinemia is associated with similar clinical and biochemical findings initially, but glycine concentrations normalize between 2 and 8 weeks of life, and the prognosis is generally more favorable.10
Treatment of NKH has focused on three compounds: (a) glycine, (b) benzoate, and (c) carnitine. Glycine is known to stimulate the N-methyl-D-aspartic acid (NMDA) receptor, and dextromethorphan is a noncompetitive agonist of this same receptor, which has been used with some clinical success. High doses of benzoate can lower the CSF concentration of glycine, and there are anecdotal reports of responses. Carnitine deficiency has been documented in patients treated with benzoate,
and therefore supplementation with L-carnitine has been advocated in patients receiving benzoate. Despite prompt administration of these treatments, some children may still suffer lasting neurologic sequelae.8
and therefore supplementation with L-carnitine has been advocated in patients receiving benzoate. Despite prompt administration of these treatments, some children may still suffer lasting neurologic sequelae.8
Table 1 Metabolic disorders in late infancy | ||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||
Table 2 Metabolic disorders in the neonatal period | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
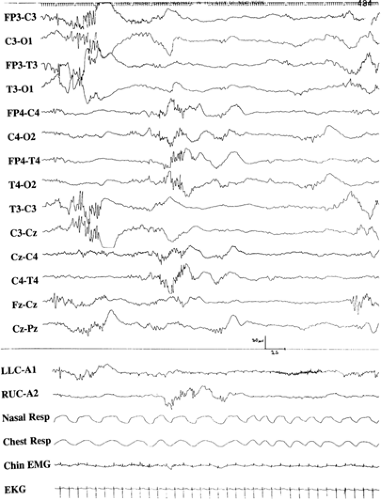 FIGURE 1. Suppression-burst electroencephalogram with interhemispheric asynchrony in a newborn. The sup- pression-burst pattern is characteristic of a variety of disorders, including nonketotic hyperglycinemia, phenylketonuria, maple syrup urine disease, and molybdenum cofactor deficiency. EKG, electrocardiogram; EMG, electromyogram; Resp, respiration. |
Pyridoxine-dependent Epilepsy (#266100)
This disorder, transmitted as an autosomal-recessive trait, was previously believed to be due to a defect involving glutamic acid decarboxylase, but linkage studies have excluded this enzyme and implicated other pathways that do not appear to be primarily involved with this reaction.5 Instead, mutations in an enzyme, antiquitin, in the lysine degradation pathway are responsible for the condition. An accumulated substrate inactivates pyridoxal 5-phosphate by forming a Knoevenagel condensation. The diagnosis can be suspected by finding elevated pipecolic acid in the blood, urine, or CSF.41
Typically, recurrent and long-lasting seizures occur in the neonatal period that are refractory to treatment with conventional antiepileptic drugs. The seizure types vary, and include persistent partial seizures with variable preservation of consciousness, recurrent status epilepticus, generalized myoclonic and atonic seizures, and infantile spasms. Progressive irritability, restlessness, and vomiting often precede seizures. The EEG may be diagnostic, showing an unusual paroxysmal pattern consisting of bursts of diffuse but asynchronous high-vol- tage delta activity intermixed with spikes or sharp waves. Other EEG findings include focal and multifocal spikes, bursts of generalized delta waves, and paroxysmal complexes of sharp and slow waves.40 Intravenous administration of pyridoxine usually produces rapid control of seizures, but some cases may require several days of oral administration to show clinical effect. Patients remain dependent on pyridoxine supplementation to maintain seizure control and normal neurologic development.
Atypical cases of pyridoxine dependency have been described. Onset may be delayed into early infancy. As a rule, all infants of this age who have idiopathic seizures should be given a test dose of pyridoxine (75–100 mg) to determine responsiveness. Occasionally, a patient with vitamin B6–dependent conditions will become apneic after the test dose is administered, and so it is important to anticipate the need for transient respiratory support. Decreased concentration of γ-aminobutyric acid (GABA) in the CSF is the defining biochemical abnormality. This finding, coupled with elevated CSF concentration of glutamate and response to administration of pyridoxine, is diagnostic.
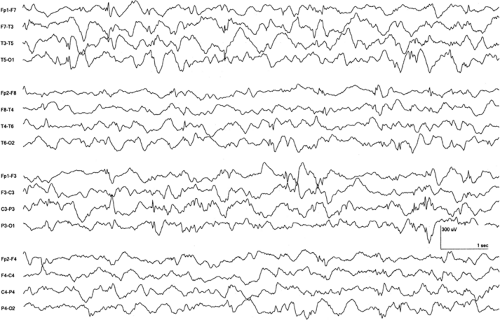 FIGURE 2. Electroencephalogram (EEG) in a patient with dihydropyrimidine dehydrogenase deficiency. The infant is 6 months old. Note the background slowing, the asymmetry with reduced complexity on the right, and the presence of multifocal spikes. A prior EEG had shown reduced complexity on the left, indicating that this feature presents with shifting laterality. (Courtesy of Dr. Kent R. Kelley.) |
Some patients with neonatal encephalopathies do not respond to B6 but do respond to pyridoxal phosphate.9,67 Pyridoxal phosphate is the biologically active form of the B6 vitamins, but the defect in these patients is not precisely known. Some advocate giving neonates or infants with intractable seizures and encephalopathy a test dose of pyridoxal phosphate (either 50 mg for a neonate or 30–50 mg/kg/d for infants) for 2 weeks because of these rare cases of pyridoxine resistance
and the observation that pyridoxal phosphate appears to be superior to B6 in idiopathic epilepsies.4,68
and the observation that pyridoxal phosphate appears to be superior to B6 in idiopathic epilepsies.4,68
Dihydropyrimidine Dehydrogenase Deficiency (#274270)
Dihydropyrimidine dehydrogenase (DPD) is used in the first step of the pyrimidine degradation pathway, converting uracil and thymine to β-alanine and the R-enantiomer of β-aminoisobutyric acid. This conversion primarily takes place in the liver, and DPD is the rate-limiting enzyme of the catabolism. This enzyme is absent in the brain, but it is believed that alanine may be converted from carnosine owing to the presence of tissue carnosinase in brain. There is a wide range of clinical presentation, but nearly all children present with neurologic abnormalities, including hypertonia, hyperreflexia, tremor, seizures, and developmental delay. Severely affected children present as newborns with hypertonia, stiffness, and feeding difficulties.2 Details of the EEG features and characteristics of the seizures have yet to be described. In our own patient, erratic myoclonias were present. The EEG showed bursts of semirhythmic delta and attenuation of the expected background complexity over either hemisphere with shifting laterality. Multifocal interictal epileptiform discharges were present (Fig. 2). No treatment has been shown to be effective.
Molybdenum Cofactor Deficiency (#252150)
This condition is exceedingly rare. Molybdenum is a trace element that serves as an essential cofactor for the reactions of three different enzymes: (a) sulfite oxidase, (b) xanthine dehydrogenase, and (c) aldehyde oxidase. Deficiency of molybdenum cofactor can result from mutations in four separate genes: MOCS1, MOCS2 (which codes for molybdopterin synthase), MOSC3, and GEPH (which codes for gephyrin).36 Absence of hepatic molybdenum cofactor results in a combined enzyme deficiency. A progressive encephalopathy develops in affected infants, with recurrent and refractory seizures appearing shortly after birth. Focal seizures, diffuse tonic seizures, and erratic myoclonic jerks are common. Jitteriness, abnormal cry, and intermittent irritability are other neurologic findings. Dysmorphic features, ectopia lentis, and hepatomegaly are associated somatic manifestations. The EEG is characterized by multifocal paroxysms and a suppression-burst pattern. Brain magnetic resonance imaging (MRI) demonstrates initial signal changes consistent with white matter edema, then curvilinear signal changes in the gray–white junction suggestive of hemorrhage and laminar necrosis, and ultimately multiple subcortical cystic areas scattered throughout the brain.16
Isolated sulfite oxidase deficiency is clinically and pathologically indistinguishable from molybdenum cofactor deficiency, suggesting that decreased sulfite oxidase activity is central to the clinical syndrome.59 In both conditions, the serum concentration of uric acid is abnormally low, and sulfur-containing
products are evident in the urine. Serum urate may be the more sensitive screening test.3 There is no effective treatment for this condition. Recent work suggests that sulfites might have a direct impact on glutamate dehydrogenase function, thereby reducing the adenosine triphosphate (ATP)-producing capacity of cells.75 In theory, a severe impairment of energy production could account for the MRI appearance and the burst-suppression pattern apparent on EEG tracings.
products are evident in the urine. Serum urate may be the more sensitive screening test.3 There is no effective treatment for this condition. Recent work suggests that sulfites might have a direct impact on glutamate dehydrogenase function, thereby reducing the adenosine triphosphate (ATP)-producing capacity of cells.75 In theory, a severe impairment of energy production could account for the MRI appearance and the burst-suppression pattern apparent on EEG tracings.
Peroxisomal Diseases
These disorders are invariably associated with seizures in the neonatal period. Zellweger (cerebrohepatorenal) syndrome (ZS) is the prototype of this group of conditions. Neonatal adrenoleukodystrophy and infantile Refsum disease are progressively milder presentations. Most cases of ZS are caused by mutations in the PEX1 gene, and the severity of the mutation correlates with the clinical severity, although to an imperfect degree.66 Severe encephalopathy with failure to thrive, hypotonia, hyporeflexia or areflexia, and seizures are the major features. Seizures develop in the neonatal period in 80% of patients. Seizure types include focal, myoclonic, and atypical flexor spasms. The EEG shows multifocal spikes or, less frequently, hypsarrhythmia. Dysmorphic features are distinctive and often permit diagnosis by inspection. These include a high, prominent forehead; shallow orbital ridges; high palatal arch; deformities of the external ear; micrognathia; presence of epicanthal folds; low, broad nasal bridge; and redundant skin folds of the neck. Cataracts, glaucoma, pigmentary retinopathy, optic nerve dysplasia, and amaurosis are usually present, as are hepatomegaly, polycystic kidneys, and calcific stippling of the patellae. Laboratory abnormalities include absence of peroxisomes on liver biopsy and high levels of very-long-chain fatty acids in the serum.
Disorders of the Urea Cycle
These conditions involve 1/30,000 live births.24 Except for ornithine transcarbamoylase deficiency, which is transmitted as an X-linked dominant trait, the other five urea cycle disorders are autosomal-recessive diseases. Hyperammonemia, respiratory alkalosis, absence of ketoacidosis, and decreased blood urea nitrogen concentrations are laboratory findings that strongly suggest a urea cycle disorder. The concentrations of citrulline and argininosuccinic acid in the plasma and of orotic acid in the urine help to differentiate among six different enzyme defects of the urea cycle.
Approximately 60% of infants with urea cycle defects become symptomatic within 24 to 72 hours of birth. Symptoms include progressive lethargy, vomiting, hypothermia, and hyperventilation. Seizures are not usually seen initially but are manifested later, as cerebral edema develops. The EEG is characterized by low-voltage, asymmetric delta and theta activity. A suppression-burst pattern may develop, and the duration of intervals between bursts may correlate with the degree of hyperammonemia. Brain imaging reveals cerebral edema with small ventricles, and intracranial hemorrhage has been reported. Primary treatment is dietary, with various drugs administered to lower the blood and tissue concentrations of ammonia.
Maple Syrup Urine Disease (#248600)
There are five subtypes of maple syrup urine disease (MSUD), but the classic severe neonatal form presents with poor sucking, lethargy, and coma, beginning sometime between the fourth and seventh days of life. Intermittent hypertonus and opisthotonus, gross tremor, myoclonic jerks, and repetitive flexion–extension movements of the limbs are common. It is the result of a defect of the branched-chain α-ketoacid dehydrogenase complex, and mutations in at least four different genes can cause the clinical syndrome. This mutation is expressed as an autosomal-recessive trait, with an estimated prevalence of 1/200,000 live births. The branched-chain amino acids (leucine, isoleucine, and valine) accumulate together with the α-ketoacid derivatives. Clear focal seizures, seizures with diffuse clinical expression, and uncontrolled cerebral edema are common in untreated subjects. A mu-like rhythm characterized by bursts of 7- to 9-Hz spindle-like sharp waves of moderate amplitude is distinctive.18 Fast rolandic rhythms have also been described.
Organic Acidurias
The disorders of organic acid metabolism comprise a large number of inborn errors, including isovaleric aciduria and several ketotic hyperglycinemic syndromes (propionic acidemia, methylmalonic acidemia, and β-ketothiolase deficiency).
Symptoms develop during the neonatal period in approximately 50% of children who have isovaleric aciduria (#243500), with poor feeding, vomiting, dehydration, and a progressive encephalopathy manifested by lethargy, tremors, seizures, and coma. Cerebral edema is present, and seizures are most often focal motor or diffuse tonic posturing. The EEG shows dysmature features during sleep. Distinctive biochemical findings include metabolic acidosis, ketosis, lactic acidosis, hyperammonemia, and transient bone marrow suppression. Isovalerylglycinuria is diagnostic. The disorder is due to a defect in isovaleryl CoA dehydrogenase. It is of note that recent newborn screening has led to the identification of asymptomatic individuals with identical genetic mutations. This is important information for genetic counseling.44
The symptoms of propionic acidemia (#606054) also appear during the neonatal period, and 20% of affected newborns have seizures as the first symptom. The defect is in propionyl-CoA carboxylase. Again, both focal seizures and manifestations that appear more diffuse have been reported. The EEG shows diffuse delta wave activity, with generalized or focal temporal spikes during the encephalopathic phase. Intractable epilepsy may develop. In 40% of affected children, generalized convulsions and myoclonic seizures develop in later infancy, and older children may have atypical absence seizures. A recent workshop showed that modern mortality rates are about one in three, but the degree of dietary protein restriction used as treatment varies considerably among centers worldwide.65 Biochemical findings include metabolic acidosis, ketosis, and elevation of branched-chain amino acids and propionic acid.
Methylmalonic acidemia is the metabolic signature of several biochemically distinct entities, all of which show decreased activity of methylmalonyl-CoA mutase.48 Stomatitis, glossitis, developmental delay, failure to thrive, and seizures are the major features. Diffuse tonic postures and focal seizures with apparent secondary generalization are the most frequently described seizure types. Methylmalonic acidemia also occurs in association with homocystinuria. Seizures involve repetitive clonic eyelid blinking and simultaneous upward deviation of the eyes. Lesions of the globus pallidus on computed tomography (CT) or MRI are classic.57
Defects of carnitine palmitoyltransferase types I and II may be manifested in the newborn period by diffuse neurologic signs and seizures.39 Hypsarrhythmia has been described in one case. Deficiency of carnitine acylcarnitine translocase also may produce seizures, apnea, and bradycardia in the neonatal period, and seizures may occur in other defects of fatty acid oxidation.28
Pyruvate Dehydrogenase Deficiency (#312170) and Pyruvate Carboxylase Deficiency (#266150)
The clinical manifestations of pyruvate dehydrogenase (PDH) deficiency are extremely heterogeneous. The spectrum ranges from neonatal lactic acidosis with severe neurologic dysfunction to a slowly progressive, chronic neurodegenerative disorder.6 Structural abnormalities, such as agenesis of the corpus callosum, are frequently revealed by brain imaging. Mutations most often affect the E1 α subunit gene located on the short arm of chromosome X.11 The pyruvate dehydrogenase complex catalyzes the irreversible conversion of pyruvate into acetyl-CoA. The PDH complex is composed of multiple copies of three enzymes: E1 (PDHA1), dihydrolipoyl transacetylase (DLAT or E2), and dihydrolipoyl dehydrogenase (DLD or E3). The E1 enzyme is made up of two α and two β subunits. The E1 α subunit contains the active site and plays a key role in the function of the PDH complex. Complex malformations of the nervous system are common in girls with neonatal onset, and seizures are a frequent feature, including infantile spasms and myoclonic seizures. Electroencephalographic abnormalities are usually severe and include multifocal slow spike-and-wave discharges. Patients with defects in E2 have been recently reported. The clinical expression is quite different, and affected individuals may present with episodic dystonia and few or none of the classical features of PDH deficiency.26 Patients are treated with the ketogenic diet because use of ketone bodies for oxidative metabolism bypasses the primary defect. A zebrafish model was developed that demonstrates the effectiveness of this approach.60
Pyruvate carboxylase deficiency also may be devastating in the neonatal period, being associated with severe lactic acidosis, hypotonia, failure to thrive, and seizures.11 The combination of lactic acidosis and ketosis is a distinctive metabolic disturbance, and when hyperammonemia, citrullinemia, and hyperlysinemia are also present, it is diagnostic of the disorder. Seizures are related to the associated hypoglycemia and failure of the Krebs cycle. Treatment with adrenocorticotropic hormone (ACTH) can worsen infantile spasms associated with this disorder, and treatment with the ketogenic diet can be lethal.52
Other disturbances of mitochondrial function have been linked to Leigh syndrome (LS) and Alpers syndrome. Symptoms of these two syndromes, which appear in infancy, include convulsions. Leigh syndrome is better understood and may be related to various biomolecular defects.14 Maternally inherited Leigh syndrome (MILS) is frequently associated with convulsions and pigmentary retinopathy. The common mutation associated with MILS is a thymine–guanine transition at nucleotide position 8993 in complex V of the mitochondrial genome. Other mitochondrial DNA mutations may produce the MILS phenotype. Leigh syndrome associated with deficiency of pyruvate dehydrogenase is characterized by convulsions about 50% of the time. In contrast, convulsions occur infrequently (7% of cases) in LS associated with cytochrome c oxidase.
The pathophysiology of Alpers syndrome is more obscure. This condition is transmitted as an autosomal-recessive trait and is invariably associated with convulsions. Liver failure with cirrhosis follows in the Huttenlocher variant.27 Whether Alpers syndrome is mitochondrial in origin had been a subject of debate.24 Mutations in the POLG gene have been reported.45
Disorders of Carbohydrate Metabolism
Hypoglycemia in the neonatal period is often caused by an inborn error of gluconeogenesis, such as deficiency of fructose-1,6-bisphosphatase. Fifty percent of affected children show symptoms during the first week of life, including periodic hyperventilation, hepatomegaly, irritability, apnea, somnolence, and coma. Other patients become symptomatic during the early infantile period. Seizures are common, and the initial EEG reveals diffuse slowing and low-amplitude background activity with intermittent bursts of fast activity. Less frequently, there are multifocal sharp waves and spikes. Profound hypoglycemia, lactic acidosis, ketosis, elevated plasma concentrations of alanine, and presence of abnormal urinary organic acids with glycerol and glycerol-3-phosphate are characteristic biochemical findings. The diagnosis can be made using cultured lymphocytes, thereby eliminating the need for a liver biopsy.31 Neurologic symptoms and permanent brain damage can be avoided by preventing hypoglycemia.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


