INCIDENCE AND PREVALENCE
Inborn errors of metabolism are individually rare. Most data in the past have relied on studies of prevalence, which are highly dependent on ascertainment of cases. This has tended to bias such numbers in favor of more severe cases, particularly those first presenting in children. In more recent years, the availability of disease-modifying therapies for many of these disorders has led to pressure for newborn screening to be performed. In the Unites States, many states now screen for 50 or more diseases, most of which qualify as inborn errors of metabolism. The institution of newborn screening has given greater insight into the birth incidence of these diseases. In some cases, it has become apparent that preexisting estimates were far too low. For example, it was thought that the birth incidence of Fabry’s disease was approximately 1 in 50,000. A study that screened more than 30,000 consecutive newborn males in the Piedmont region of Italy found that the birth incidence was closer to 1 in 3,000. Of note, the majority of these cases were predicted to be late onset, as opposed to the classical childhood onset form of the disease. More widespread application of such screening has confirmed that previous figures were too low and that it is likely that the majority of cases of inborn errors of metabolism are actually those with milder phenotypes, which may go unrecognized or misdiagnosed. Thus, it is incumbent on all neurologists, including adult neurologists, to be familiar with these disorders, which can present at any age and in many guises.
CLASSIFICATION
The complexity of metabolic pathways in the cell is such that mutations in any one gene may lead to a large variety of downstream consequences. Any classification is thus an oversimplification. Nevertheless, for practical purposes, a division of inborn errors of metabolism into small and large molecule diseases is clinically useful.
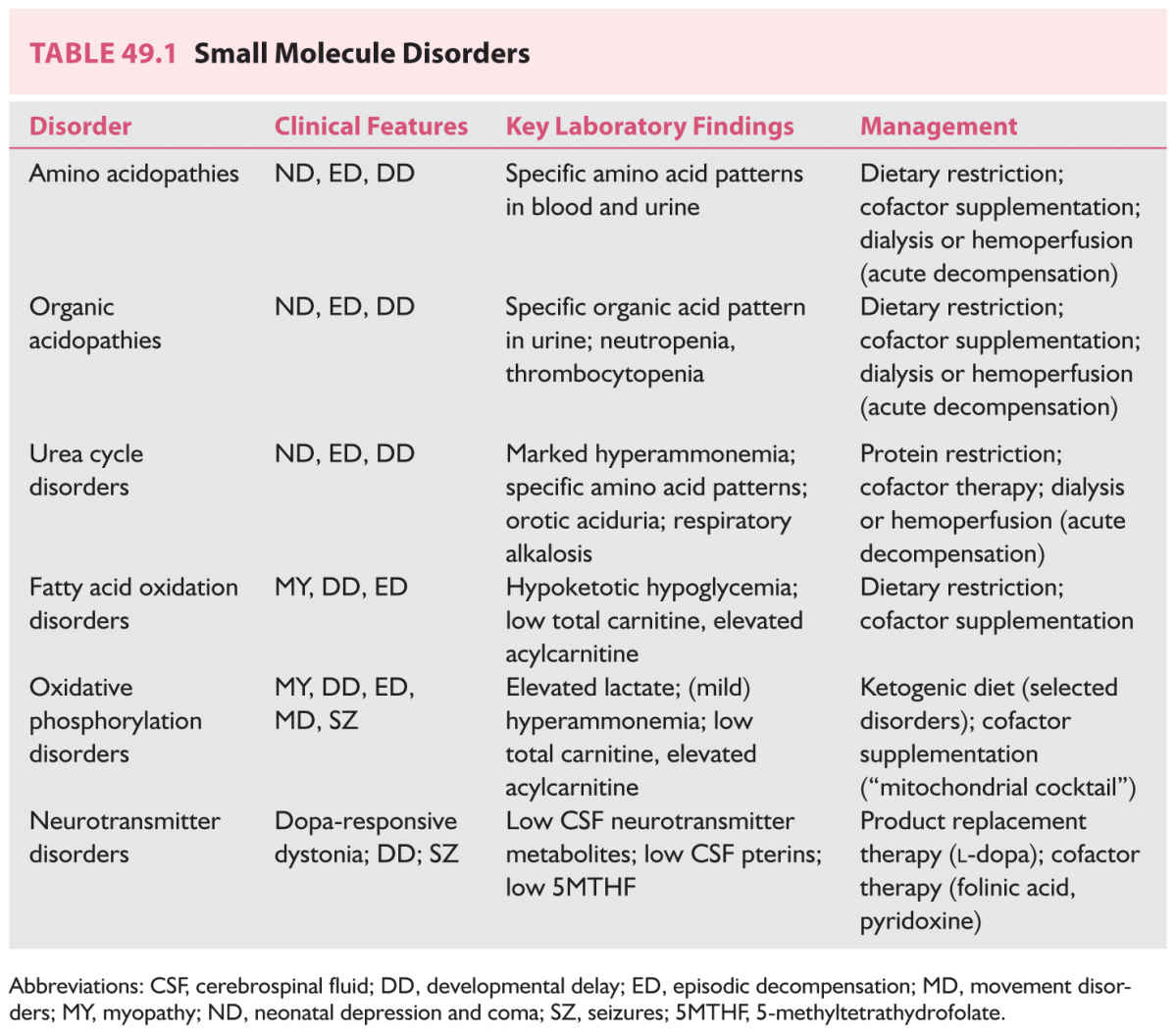
The small molecule diseases are those involving amino acids, organic acids, the urea cycle, the respiratory chain and the synthesis of neurotransmitters, among others. In their most severe form, these disorders present in neonates or young children, in whom the deficiency of the enzyme or other gene product is so profound that these children are unable to metabolize even normal substrate loads. They will often present with catastrophic deterioration in the newborn period when they are first exposed to a normal substrate load, typically when they begin to feed.
Alternatively, those with less severe loss of activity of the gene product may present only when they are confronted with an increased substrate load, such as that which occurs in a hypercatabolic state associated with fever, or in association with a change in diet. Common presenting features may include coma, seizures, acidosis or movement disorders. There may be evidence of multiorgan failure in many of these phenotypes.
A prime example is the X-linked disorder, ornithine transcarbamylase deficiency, the most frequent of the urea cycle disorders. In newborn males (hemizygotes), this presents after first exposure to a protein load with coma and cerebral edema; mortality is high, even with timely and aggressive intervention. In heterozygotes (female carriers), presentation may be delayed until adolescence or adulthood, when women may present with migrainous features or stroke-like episodes. Pregnancy, the greatest metabolic stress experienced by normal women, is often the precipitating event.
Some small molecule diseases may mimic large molecule diseases. The cerebral organic acidemias are a good example of this. In many of these, there is a slowly progressive presentation with or without typical imaging findings. Examples include glutaric aciduria type 1, and succinic semialdehyde dehydrogenase deficiency. Affected individuals may also decompensate in the face of intercurrent infection or increased substrate loads.
The small molecule diseases may be conveniently classified as shown on Table 49.1. Typical presentations and abnormal substrates for analytic purposes are also shown here.
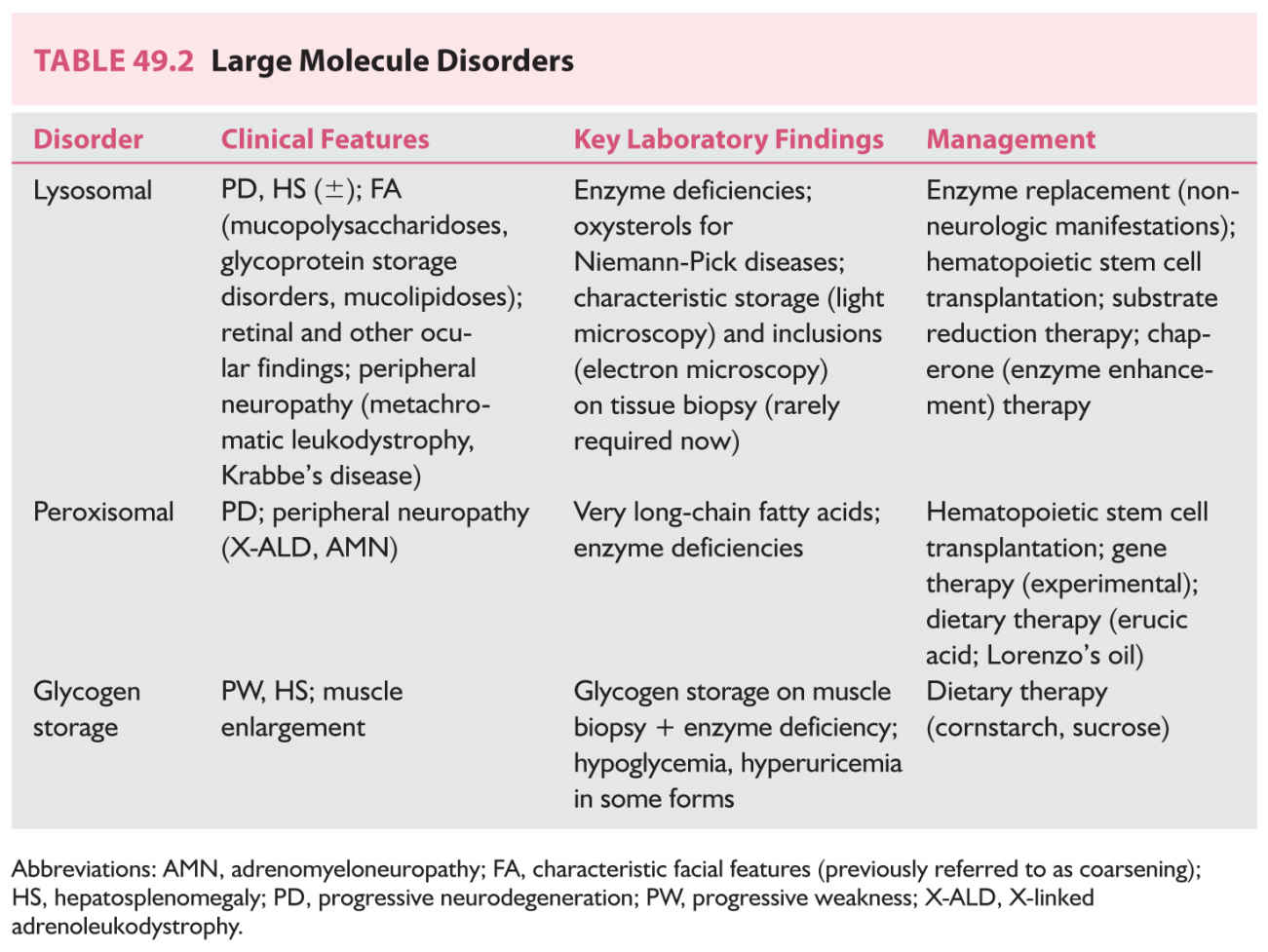
These are the disorders typically described as “storage diseases.” They usually have an insidious, slowly progressive presentation in which what is initially thought to be intellectual disability may eventually declare itself as dementia. Organomegaly and changes in the eyes, skin, hair, and nails are commonly seen, but need not be present to diagnose one of these disorders. They are usually classified based on the biochemistry of the accumulating substrate. In addition, the organelle to which they are localized is used as part of an overarching classification. Thus, lysosomal storage diseases, first characterized in the 1950s, when the ultrastructural anatomy of the cell first began to be appreciated, are subclassified as sphingolipidoses (disorders in which there is excessive storage of lipids), mucopolysaccharidoses (complex polymers of sugars accumulate), neuronal ceroid lipofuscinoses (proteins accumulate in excess), mucolipidoses (overlapping features of sphingolipodoses and mucopolysaccharidoses) and glycoprotein storage diseases (glycans [glycosylated proteins] accumulate in excess). Peroxisomal disorders are classified according to the nature of the deficiency of the gene product; elevated very long-chain fatty acids are characteristic. In the classic archetype, the Zellweger’s syndrome, unusual facial features, organomegaly, bone abnormalities (chondrodysplasia punctata) and neurodegeneration are characteristic. The Zellweger phenotype is in fact a family of diseases in which mutations involving different transport proteins (peroxins) lead to deficiency of multiple enzymes within the peroxisomal matrix. In other disorders, a single gene product may be defective. Thus, X-linked adrenoleukodystrophy, commonly presenting in boys in middle childhood with various agnosias, spasticity and rapid neurologic deterioration, result from deficiency of a single enzyme transported into the peroxisome by the ALD protein.
Table 49.2 summarizes the findings in these disorders.
Some disorders defy easy categorization into small and large molecule disorders. The rapidly expanding family of congenital disorders of glycosylation (CDG; more than 90 types at the time of writing) comprise such a group. The most prevalent type, comprising about 80% of all CDG cases, is phosphomannosmutase 2 (PMM2) deficiency, which features episodic metabolic decompensation, stroke-like episodes and seizures in infancy and early childhood, but a more slowly progressive, large molecule disorder-like course in those who survive to the second and later decades. Similar diversity is encountered among the other N- and O-linked disorders of glycosylation, ranging from nonsyndromic intellectual disability in TUSC3-CDG, to congenital muscular dystrophy phenotypes (Walker-Warburg syndrome, Fukuyama congenital muscular dystrophy) to severe neurologic phenotypes with essentially no development and intractable seizures (ALG8-CDG).
DIAGNOSIS
The most important element in making a diagnosis of an inborn error of metabolism affecting the nervous system is a high index of suspicion. Any individual presenting with characteristic features of these phenotypes should be investigated. However, we increasingly recognize that late-onset diseases may have fragmentary or atypical phenotypes. Inborn errors of metabolism should be included in the differential diagnosis of any neurologic disorder. When the phenotype is typical, diagnostic testing may be focused to a specific enzyme, substrate, or gene. However, in cases presenting in more nondescript fashion, such as children with developmental delay with or without hypotonia and other suggestive features, or adults with early-onset dementias or treatment-resistant psychiatric illness, more broad screening may be appropriate. It is important to remember that screening tests have false negatives and false positives and, particularly in the case of small molecule diseases, that the ideal time to obtain a sample is when the patient is most symptomatic. Unfortunately, this commonly seems to occur after hours when regular services may not be available. Thus, it is important to obtain, collect and transport such samples in such a fashion that they will be in suitable condition for analysis. If there is any suspicion of an inborn error of metabolism in an adult or child who has decompensated, it is prudent to discuss the case with the biochemical genetics laboratory to ensure that appropriate samples are obtained.
Typical screening profiles will include quantitative plasma amino acids, urine organic acids, plasma lactate, pyruvate and ammonia, serum carnitine profile, complete blood count and biochemical profile plus blood gas analysis. Such investigations will enable diagnosis of most of the amino acid, organic acid, and urea cycle disorders and may provide clues to the diagnosis of a respiratory chain or fatty acid oxidation disorder. Confirmation of the diagnosis is by sequencing of the relevant gene (individually or in a panel) in most cases; this information is important for prognosis and allows for antenatal diagnosis for couples who wish to avail themselves of this option.
More specialized testing is required in the case of neurotransmitter disorders in which cerebrospinal fluid must be obtained and rapidly cooled on dry ice, then promptly shipped to a reference laboratory. When obtaining cerebrospinal fluid (CSF), it is important to measure CSF glucose with simultaneous serum glucose to permit appropriate interpretation. In addition, any time that the cerebrospinal fluid glucose is measured, the lactate and pyruvate should also be measured. Disorders with hypoglycorrhachia and increased CSF lactate may result from bacterial infection or respiratory chain disorders. On the other hand, a low (less than 40% of serum glucose) CSF glucose and low CSF lactate point strongly to glucose transporter type 1 deficiency, a disorder readily treatable with the ketogenic diet in children.
A child with weakness attributed to muscle disease who presents with decompensation and hypoglycemia without ketonuria may have a disorder of fatty acid oxidation. Assay of serum fatty acids, a carnitine profile, acyl carnitines and acyl glycines can help round out the evaluation of such a child and are likely to detect disorders of fatty acid oxidation and many respiratory chain disorders. Urine screening is helpful for disorders of creatine synthesis, complemented by magnetic resonance spectroscopy of the brain.
Urine screens for purine and pyrimidine disorders may be helpful in patients presenting with delay or autistic features, particularly if they are associated with megaloblastic anemia or hyperuricemia.
In patients with slowly progressive loss of neurologic function (cognitive decline, progressive spasticity, weakness, movement disorders) particularly with evidence of systemic storage, the urine may be screened for mucopolysaccharides and oligosaccharides; false negatives may occur with some of these disorders. More sophisticated mass spectrometry techniques have improved the sensitivity and detection ability of urinalyses for these conditions. Direct assay of one or more lysosomal enzymes in white cells, serum, or fibroblasts is often employed when these disorders are suspected, although in certain cases (e.g., in at-risk populations with a high frequency of specific mutations), direct molecular analysis may be appropriate.
As next-generation DNA-sequencing technology continues to fall in cost, and as software algorithms for analysis of the massive data produced by these techniques are refined, whole-exome, and eventually, whole-genome sequencing will eventually become first-line screening and diagnostic techniques for inborn errors of metabolism. Because these methods find many DNA sequence variants whose pathogenicity is unclear, sophisticated biochemical analytical techniques will not be supplanted, since these will be critical in establishing the functional significance of DNA variants.
TREATMENT
In general, the small molecule disorders have a wider range of treatment options than the large molecule disorders. The prototype of these disorders is phenylketonuria, in which a phenylalanine-restricted diet applied early, and maintained rigorously, clearly improves the outcome. Since newborn screening for this disorder was instituted a half century ago in the United States, the prevalence of individuals impaired by phenylketonuria has dropped dramatically. It has been learned, however, that dietary restriction must be maintained at least through the adult years and perhaps lifelong, since failure to adhere to the diet can lead to recurrence or the development of new neurologic symptoms. Many of the amino acid and organic acid disorders can be managed with specific diets that qualify as medical foods. In some cases, the administration of specific cofactors may be highly beneficial. Thus, some forms of homocystinuria may respond to cobalamine and others to pyridoxine, depending on the precise enzymatic deficiency. Biotinidase deficiency, which can present with developmental delay, dermatitis and myoclonic seizures, with or without abnormal organic acids in the urine, responds well to the administration of pharmacologic doses of biotinidase. Disorders for which disease-modifying therapy is of proven value, and is readily available, should take priority in diagnostic workup.
In some cases, definitive therapy may take the form of organ transplantation and gene therapy is beginning to be investigated for these disorders.
Progress in large molecule diseases has been slower, but hematopoietic stem cell transplantation, enzyme replacement therapy, and small molecule therapy all have now established places in the treatment of these disorders, particularly the so-called lysosomal storage diseases.
It is clear, even if definitive therapy is not available, that early diagnosis is beneficial to affected individuals. Not only does this mean that clinical problems may be anticipated and managed expectantly, but it also allows the diagnostic odyssey to be ended and to provide the family with useful information for family planning, should the diagnosis be made sufficiently early. The impact of newborn screening on these diseases has yet to be assessed. However, preliminary data suggest that many patients will be detected as newborns, who would not be expected to present until much later in life. How to manage these individuals both medically and from the psychosocial point of view is proving to be a challenge, which has not yet been fully met.
PREVENTION
Given difficulties in diagnosis and continuing limitations in disease-modifying therapies, the ideal approach to inborn errors of metabolism is to prevent their occurrence. The greatest success story in this regard is the carrier screening program for Tay–Sachs disease. Tay–Sachs disease is a form of GM2 gangliosidosis, inherited in autosomal recessive fashion, which classically presents in infants with developmental stagnation and regression associated with hypotonia, macrocephaly, seizures and an exaggerated startle response. Children typically have cherry-red spots at the macula. This disease has a carrier frequency in the Ashkenazi Jewish population of approximately 1 in 30. After the enzymatic deficiency was discovered in 1969, it was found that carriers could be identified by screening blood tests and a prevention program was established. This has proven to be an enormous success, whose widespread implementation led to a dramatic reduction in the incidence of Tay–Sachs disease in the Ashkenazi Jewish population. Indeed, most cases of Tay–Sachs disease are now seen in non-Ashkenazi subjects.
The availability of a simple screening test, and a high carrier frequency in the at-risk population were key factors in the success of this approach. These factors are not present in many other inborn errors of metabolism. However, progress in DNA-sequencing technologies, particularly the widespread application of next-generation sequencing techniques, has raised the possibility of broad screening for rare diseases on a population basis. There are still many practical, ethical and social issues to be addressed before this can be implemented, but this new technology does hold the promise of eventually dramatically reducing the incidence and prevalence of these diseases, which are individually devastating and collectively costly.
Key Points
• Inherited metabolic neurologic disorders are individually rare, but collectively common, owing to the large (and growing) number of such disorders.
• Neurologists must be familiar with these disorders because they may present at any time during the life span, because many have disease-modifying therapy available, and because accurate and timely diagnosis ends what is often a prolonged diagnostic odyssey, allows the neurologist to intervene appropriately, and provides data for prognostication and counseling.
• Although no classification is entirely satisfactory, grouping these disorders into small and large molecule disorders is of practical value.
• Small molecule disorders typically result from enzyme deficiencies. Complete deficiency results in presentation in the newborn, when first exposed to a substrate load, often the first feeding. Less profound deficiencies result in episodic presentations in older children, or even adults, where the residual enzyme activity is sufficient for everyday demands, but decompensates in the face of intercurrent infection, pregnancy, or other hypercatabolic states.
• Samples of blood, urine, or CSF for metabolic testing should be obtained when the patient is acutely ill; normal assays when the patient is well do not rule out small molecule diseases.
• Large molecule disorders are the classic “storage” disorders, in which macromolecules accumulate in specific tissues, leading to distinct phenotypes.
• Patients with large molecule diseases may present with
• Developmental delay
• “Cerebral palsy”—with progressive physical findings
• Epilepsy, particularly if intractable
• Psychiatric symptoms, often refractory to therapy
• Early-onset dementia
• All of the above with varying combinations of somatic findings—organomegaly need not be present to diagnose “storage” disorders.
• All patients with inherited disorders benefit from accurate and timely diagnosis, and all are helped by treatment, even if specific disease-modifying therapy is not yet available.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


