(1)
Asheville, NC, USA
Neurodegenerative disorders are diseases of the nervous system in which neurons deteriorate and die off and neural circuits become disabled. The best known of these are Alzheimer’s disease and Parkinson’s disease. These diseases affect millions of individuals worldwide. Some of these disorders have been known since ancient times. Description of symptoms typical of Parkinson’s disease appear in ancient Indian and Chinese medical texts, and then again in classical Greek and Roman writings. It was described by Galen (138–201) and by Zhang Zihe (1156–1228) in his classic Ru Men Shi Qin. Parkinson’s symptoms were described neurologically for the first time by the English surgeon James Parkinson (1755–1824). In his 1817 paper entitled “An essay on the shaking palsy”, he described its main symptoms—tremulous motion, reduced muscle strength, and bent posture. A far-more complete clinical description of Parkinson’s disease including an increased emphasis on slow movement as a major clinical feature was provided by great French neurologist Jean-Martin Charcot (1825–1893) widely regarded as a father of modern neurology. He named the disorder Parkinson’s disease, and in his famous 1872 lectures carefully delineated differences between it and other movement disorders.
The “term movement disorder” encompasses a broad spectrum of neurodegenerative diseases that produce unwanted or hyperkinetic movements such as tremors. Included in this grouping are both Parkinson’s disease and Huntington’s disease. The former is strongly identified with failing dopaminergic neurons located in the substantia nigra region of the midbrain while Huntington’s disease involves neurons located throughout the brain with some regions impacted more than others. Huntington’s disease is named for the American physician George Huntington (1850–1916) who described this disease in 1872. In his paper, he used the term chorea, taken from the Greek choreia, which means to dance, and refers to the irregular and spasmodic muscle movements that first signal the presence of the illness. The use of that term was not new, but rather was first applied to Huntington’s disease by the Renaissance physician and founder of the field of toxicology, Paracelsus (1493–1541).
Huntington’s disease has an interesting if not outright disturbing history in the United States and Europe. It was the subject of a largely discredited 1916 paper by Charles Davenport. In his paper, he connected the entry of this disease in this country to three families that migrated in the seventeenth century from the village of Bures in Suffolk County, England to Connecticut. He then linked members of these families to women charged with practicing witchcraft. This theme was later amplified in a notorious 1932 paper by Percy Vessie that asserted that people suffering from Huntington’s disease were prone to criminality and other forms of social misconduct. These arguments were subsequently amplified and used as a justification for forced sterilization and restricted immigration as part of the eugenics movement that flourished for 40 years or more in the United States and Europe.
One of the main characteristics of Parkinson’s disease, Huntington’s disease, and other forms of neurological disorder is their increase with age. That there is a connection between aging and cognitive decline is not a new concept. It can be traced back to Pythagoras who noted in the seventh century B.C. that mental decline takes place during the later stages on life. According to Solon (c.630 B.C. to c.560 B.C.), the Athenian law giver and statesman, a person’s mental condition has a bearing on the legality of late-life alterations to a will. Galen referred to late-life mental decline as “morosis”, and thought of old age as a disease condition in itself. Galen was an accomplished anatomist, and performed numerous dissections of monkeys and pigs, human dissections being prohibited. His tract became the standard medical text for the next 1400 years. It was eventually updated and supplanted by Paracelsus and Andreas Vesalius (1514–1564), who was able to use human cadavers in his anatomical investigations.
The emergence of neurological disorders as diseases having underlying physical causes took considerable time. For a long time women suffering from movement disorders were regarded as witches, while individuals undergoing late-life mental decline were often treated as criminals and locked up in prisons or asylums (Fig. 1.1). The term “cognitive” refers to tasks carried out by the brain involving knowing, thinking, remembering, organizing, and judging. However, rather than applying the term “cognitive decline” to late-life neurological disorders the far-more pejorative designation “senile dementia” was assigned. This was an exceptionally broad term due in large measure to the inability of anatomical investigations to discriminate and pinpoint physical correlates of the symptoms being exhibited. Senile dementia was taken to encompasses forms of madness such as the ones depicted by Shakespeare in Hamlet and King Lear. At other times it included illnesses such as syphilis and schizophrenia. The term “hardening of the arteries” was applied to the syndrome, while individuals showing similar signs earlier than age 65 were regarded as suffering from an early-onset dementia, or dementia praecox.
Fig. 1.1
Treatment of dementia through the years. (a) Left-hand panel: Woodcut depicting a victim of a witch trial. (b) Right-hand panel: Philippe Pinel, a champion of the humane treatment of the mentally ill, shown at the infamous Bicêtre asylum (from Berchtold Neurobiol. Aging 19: 173 © 1998 Reprinted by permission from Elsevier)
Substantial progress in cataloging and discriminating between the various types of brain disorders began with the work of early pathologists such as the British physician William Cullen (1710–1790). He introduced the term “neurosis” meaning a nervous disorder and medical condition, and included senile dementia under that heading. Further steps were taken during the nineteenth century. These included the advent of more humanitarian conditions for the treatment of the mentally ill and improvements in clinical diagnosis. The notion of age-related dementia was refined by Samuel Wilks (1824–1911), who noted in 1864 that anatomical changes (atrophy) in the brain accompanied senile dementia. The decline in brain vasculature was noted and these observations produced the leading theory of senile dementia at the end of the nineteenth century—the decline in blood flow to and in the brain results in a loss of brain oxygenation and this was the cause of senile dementia.
Alzheimer’s disease (AD) is the leading cause of senile dementia. It is estimated that 13 % of the people over the age of 65 suffer from AD. It is considerably worse for people over the age of 85—for them the likelihood of exhibiting Alzheimer-like symptoms increases to 45 %. The statistics are indeed grim. Worldwide, 5 million new cases of AD are diagnosed every year (or one new case every 7 s). Given the attention that this illness now receives it is perhaps surprising how little attention it got in 1906 when the German pathologist Alois Alzheimer (1864–1915) carried out his groundbreaking work. At that time the (juvenile) illness he had diagnosed was regarded as a new illness, a form of presenile dementia having only the most tenuous connection to the more common senile dementia seen in elderly individuals. That situation gradually changed with the realization that most of the cases of senile dementia had the same histopathological signs as the condition described by Alzheimer, namely, extracellular senile plaques and intracellular neurofibrillary tangles.
1.1 Alzheimer Discovers Amyloid Deposits in the Brain
In order to make progress past the anatomical level of observation, and distinguish between different kinds of neurological disorders, greater resolution of brain tissue was needed. That was provided by the light microscope. Invented by the Dutch lens-maker Anton van Leeuwenhoek (1632–1723) and improved upon by the English scientist and inventor Robert Hooke (1635–1703). This device enables the user to observe cells, and their organelles, and other macromolecular substructures. Contrast is important, and advances in the field have occurred whenever new and improved dyes have been discovered that make the macromolecular structures of interest stand out against the cellular background. The amount of detail made visible by a light microscope is limited by the wavelength of the probe being used. When used with the right dyes the light microscope can resolve structures down to about 0.2 μm, or 200 nm (half the 400 nm nominal wavelength of visible light).
The light microscope is the primary tool of the pathologist. This field has its modern beginnings in the pioneering studies of Rudolf Virchow (1821–1902) in the mid-nineteenth century. Virchow is regarded as the father of modern pathology. In 1854, he applied the term “amyloid” to describe unusual deposits found in the liver, the spleen, and other organs and tissues of people who had succumbed to illnesses ranging from tuberculosis to syphilis. These deposits were initially thought to be fatty or waxy in nature; the term amyloid had been coined to a few years earlier, in 1838, by the botanist Mathias Schleiden in his description of starch and cellulose. The correct identity that these deposits were proteinaceous was made a few years after Virchow’s initial discoveries.
The first steps in discriminating between the different kinds of neurological disorders were taken by Charcot who combined careful clinical observations with anatomical studies and tissue pathology. He was the first to describe amyotrophic lateral sclerosis (ALS). In his lectures delivered at the Hôpital de la Salpêtrière in Paris, he distinguished between upper and lower motor neurons and began the process of separating the different motor diseases from one another. In the United States, this disorder is referred to as Lou Gehrig’s disease after the famous baseball player who died from it in 1941, but on the Continent and elsewhere it is known as Charcot’s disease.
The next and arguably the most famous in a series of landmark events was the 1906 presentation by Alzheimer (Fig. 1.2). In it, he described his postmortem analysis of brain tissue taken from Auguste Deter, a 51-year-old patient (in 1901) suffering from early-onset dementia. Using a light microscope, and utilizing the newly developed Golgi silver staining techniques (see Appendix 1), Alzheimer found that the tissue was permeated with extracellular deposits that he called “miliary foci” and are now referred to as extracellular plaques. He also observed that the cells themselves contained dense bundles of fibrils. He then made the conceptual leap that these deposits were in some manner causally connected to the patient’s dementia.

Fig. 1.2
Alzheimer’s research group at the Royal Psychiatric Clinic of the University of Munich in 1910. Lewy is standing at the far right and Alzheimer is third from the right in the upper row (from Goedert Nat. Rev. Neurol. 9: 13 © 2013 Reprinted by permission from Macmillan Publishers Ltd)
1.2 Amyloids Are Discovered Elsewhere in the Brain and in Different Tissues of the Body
Alzheimer’s findings were not unique to the dementia he was exploring, or the story would end there. Instead, his discoveries were preceded by those of Andrew Pick (1851–1924) who reported in 1892 that deposits were present in brain tissue of deceased patients suffering from senile dementia. These aggregates are now known as Pick bodies, and these deposits along with an extreme atrophy in the frontal and temporal regions of the brain characterize the dementia. Clinically the disorder observed by Pick differs from Alzheimer’s disease. In the former case, social and personality deficits predominate in the early stages whereas in the latter instance memory loss occurs first. A few years after Alzheimer’s report, in 1912, Frederic Lewy (1885–1950) described the presence of intracellular deposits (now known as Lewy bodies ) in the brains of patients with Parkinson’s disease. Thus, it was becoming clear that deposits of some sort of protein are associated with a large number of different diseases of the brain.
The question then naturally arose as to whether the brain deposits were the same or similar to those seen elsewhere in the body by Virchow and his successors. That question was answered in the affirmative by the development of a test specific for amyloid in 1927. In that year, Paul Divry (1889–1967) and Marcel Florkin (1900–1979) published results of their studies of various body amyloid tissues stained with a dye called Congo red and viewed using polarized light. When that is done, the amyloid tissue exhibits an apple-green birefringence. In a second 1927 paper, Divry showed that the same apple-green birefringence can be seen for extracellular plaques and again in 1934 for the intracellular tangles. The dye, Congo red, used by Divry originated in the textile industry where it had been invented in 1883 and was given that mysterious-sounding name Congo red as a marketing ploy. Shortly thereafter it was tried as a potential histological stain for tissues. Although it is no longer used to stain textiles, it is still widely used to detect the presence of amyloid in the manner established by Divry, that is, in conjunction with polarized light and the revealing of apple-green birefringence patterns (Fig. 1.3).
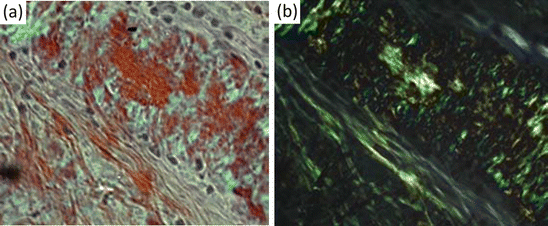
Fig. 1.3
Detection of amyloid tissue using Congo red dye (a) Congo red stained section viewed under a light microscope. (b) Same section viewed using an optical microscope and a pair of crossed polarizers showing apple-green birefringence (from Howie [from the cover] Lab Invest 88: 232 © 2007 Reprinted by permission from Macmillan Publishers Ltd)
1.3 Amyloid Structure Is Revealed Using the Electron Microscope
Light microscopes are not able to reveal the underlying structure of amyloids. They simply do not have sufficient resolution, but an electron microscope does—about three orders of magnitude greater resolution than a light microscope. The existence of birefringence patterns for these deposits makes it likely that their underlying components are arranged in an orderly fashion rather than in an amorphous manner as most people thought. In 1959, Cohen and Calkins used an electron microscope (Appendix 2) to examine the submicroscopic structure of amyloids taken from different organs of the body (spleen, liver, and kidney). They found that amyloids do indeed possess an orderly substructure. The fibrils are straight and rigid—from 75 to 120 Å in width and vary in length from 1000 to 16,000 Å (Fig. 1.4). The fibrils, in turn, are composed of protofibrils; these filamentous subunits range from 25 to 35 Å in diameter. Two or more of these units, twined about one another, form the fibril. Finally and significantly, they observed a similar structural arrangement for all the amyloids they examined.
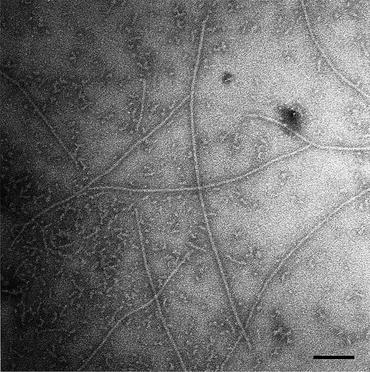
Fig. 1.4
Electron micrograph of Aβ1–42 amyloid fibrils formed in water from Aβ (1-42), and stained with 0.1 % phosphotungstic, acid showing long straight fibrils of 70–80 Å diameter within a fibrillar aggregate background. Bar = 1000 Å (from Serpell Biochim. Biophys. Acta. 1502: 16 © 2000 Reprinted by permission from Elsevier)
To summarize, by 1960 it had become apparent that amyloids deposits were associated in some as yet to be determined manner with a diverse collection of illnesses of the body and mind. These aggregates were present in extracellular spaces and within cells of various tissues and organs. Amyloids are proteinaceous aggregates characterized by: (1) birefringence patterns when stained with Congo red and viewed under polarizing light microscope conditions, and (2) composed of long-twisted fibrillar structures when examined using an electron microscope.
As is the case with any major findings in science, answering one set of questions leads to the next set of perhaps even better questions. In the case of the amyloids, the first question that arises follows from the striking similarity of the structures observed in different parts of the body. The question is: are all amyloid deposit s generated by the same protein or are amyloid deposits generated by different proteins perhaps specific to the tissues or disease states to which they are associated?
1.4 Amyloids Are Produced by Many Different Proteins
To answer this type of question, the primary amino acid sequences of the amyloidogenic proteins needed to be determined. This was accomplished for the first time in 1971 and then over the followed couple of years by several research groups starting with efforts headed by George Glenner and Earl Benditt. These investigators found that the proteins were not the same, but rather each one amyloid protein was unique to the disease associated with their presence. The primary structure, that is, the amino acid sequences, of the amyloidogenic proteins found in different tissues and organs had little resemblance to one another; they normally fold into a diverse set of 3D structures, and unlike the amyloids are soluble. The diverse set of amyloidogenic proteins, their precursors, and the disorders associated with them are summarized in Table 1.1.
Table 1.1
The systemic amyloidoses
Amyloid protein | Disease | Precursor protein | Syndrome |
|---|---|---|---|
AL/AH | Immunoglobulin light/heavy chain amyloidosis | Immunoglobulin light/heavy chains, N-terminal LC variable domain fragments | Produced by elevated clonal population of B cells; multiple organs affected; kidney, and heart failure |
AA | AA amyloidosis, Familial Mediterranean fever | Serum amyloid A (SAA) protein | Acute phase protein; concentration elevated in inflammation/injury; Affects liver, spleen, and kidneys |
ATTR | Senile systemic amyloidosis, Familial amyloid polyneuropathy | C-terminal fragments of wild-type transthyretin, mutant transthyretin | Leading cause of polyneuropathy and cardiomyopathy |
Aβ2M | Hemodialysis-related amyloidosis | β2-Microglobin | Concentration elevated in kidney disease; accumulates in bones and joints |
AApoAI | ApoAI amyloidosis | N-terminal fragment of apolipoprotein AI | Polyneuropathy and cardiomyopathy |
AApoAII | ApoAII amyloidosis | N-terminal fragment of apolipoprotein AII | Polyneuropathy and cardiomyopathy |
AApoAIV | ApoAIV amyloidosis | N-terminal fragment of apolipoprotein AIV | Polyneuropathy and cardiomyopathy |
AGel | Familial amyloidosis of Finnish type | Fragments of mutant gelsolin | Buildup in the eye, progressive polyneuropathy |
AFib | Hereditary fibrinogen amyloidosis | Mutant fibrinogen A α-chain | Renal failure; visceral, vascular, cardiac, and neurological involvement |
ALys | Lysozyme amyloidosis | Lysozyme mutants | Buildup in the viscera, especially liver, kidneys, and gut; renal disease/liver rupture |
ACys | Hereditary cystatin C amyloid angiopathy | Cystatin mutants | Buildup in tissues and cerebral arteries; cerebral hemorrhage |
The deposits of waxy materials puzzled over by Virchow in the 1850s occurred in the spleen, liver, and kidneys. They were most likely due to what is today called AA amyloidosis involving buildup of amyloid A (AA) protein over time as a consequence of long-lasting infections and chronic inflammation produced, for example, by tuberculosis and rheumatoid arthritis. Amyloid A protein deposits are derived from an overproduction of serum amyloid A (SAA), an acute phase protein serving as a precursor for the AA protein. These buildups occur most often in the heart but can occur in other tissues and organs as well as observed by Virchow.
In systemic amyloid diseases such as AA amyloidosis discussed in the previous paragraph, the amyloids collect in multiple organs and tissues of the body. In another disease of this type, immunoglobulin light chain amyloidosis (AL), B-cells secrete light chain peptides that circulate through the body and collect in the kidneys and also in the heart and liver leading to organ failure and death. Light chain amyloidosis is associated with B-cell myelomas. In contrast to the abovementioned systemic amyloidoses, there are a number of diseases in which amyloid deposit ion is specific to a single organ or tissues of the body. An example of this class of amyloids diseases is Type 2 diabetes in which aggregates of islet amyloid polypeptide (IAPP, amylin) form in the pancreas. These islet amyloid deposits of IAPP are believed to be a major contributor to β-cell failure.
1.5 Atomic Level Discovery of the Cross-Beta-Pleated Sheet Conformation
As indicated in Table 1.1 the amyloidogenic proteins are anomalous deposits built from nonamyloidogenic precursors or from fragments thereof. In order to probe further what has occurred to produce the anomalous deposits one has to examine the atomic level structure of the proteins. This requires using more powerful techniques than light or electron microscopy. The paramount experimental techniques for exploring protein structure at atomic level of detail are X-ray crystallography and nuclear magnetic resonance (NMR). These tools, and especially X-ray crystallography, were used to explore the composition of the deposits in the later 1960s and produced a breakthrough set of findings. Specifically, two sets of results were reported—one set by Eanes and Glenner in 1968 and the other by Bonar, Cohen, and Skinner in 1969. In brief, these groups reported that the amyloidogenic proteins have folded into three-dimensional forms quite different from their normal ones. In both studies, the amino acid residues are folded back and forth to form a series of β-pleated sheets. The protein chains in this configuration were then stacked in a perpendicular orientation one on top of the other along the fibril axis. A modern rendering of this type of structure is presented in Fig. 1.5.
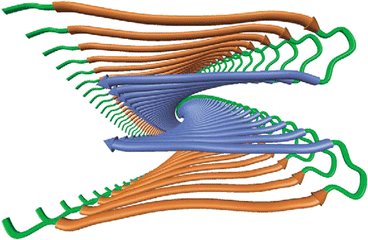
Fig. 1.5
Minimalist model of the cross-beta structure of an Alzheimer’s disease Aβ fibril as determined by means of NMR and X-ray crystallography . The view is down the long axis of the fibril (from Tycko Curr. Opin. Struct. Biol. 14: 96 © 2004 Reprinted by permission from Elsevier)
Transthyretin (TTR) is a carrier protein that transports thyroxine (T4) and retinol (vitamin A). In its amyloidogenic ATTR form, it is associated with three diseases in the elderly—senile systemic amyloidosis (SSA), familial amyloid polyneuropathy (FAP), and familial amyloid cardiopathy. This protein became the subject of a number of pioneering studies of amyloid formation. One of these was that while TTR was normally soluble, if the pH conditions in their environment were lowered, the proteins partially unfolded and then refolded into an alternate 3D conformation. In that shape, the TTR proteins became aggregation-prone, insoluble, and were capable of forming amyloid fibrils . This type of result demonstrating that the biochemical and biophysical environment has as influence on how proteins fold will be examined in greater detail in the next two chapters. Suffice it to say that at the time the results solidified the main conclusion from both the biophysics and biochemistry—normally soluble proteins have folded into a conformation different from their functionally expected one resulting in the formation of insoluble (and detergent-resistant) amyloids.
Thus, an amyloid is a material satisfying three criteria. It:
Exhibits apple-green birefringence when viewed under polarized light using an optical microscope;
Possesses a fibrillar structure when seen with an electron microscope; and
Is organized into structures composed of β-pleated sheet when examined by means of X-ray crystallography .
The linkage between these levels is a tight one—it was shown by the Glenner group in 1972 that the atomic level, beta-sheet arrangement is indeed responsible for the properties revealed by Congo red staining.
1.6 The Brain Protein Aggregates Are Characterized Biochemically and Genetically
It is interesting to note that Alzheimer’s disease was initially regarded as not being particularly important; and due to its early onset it was not regarded as the same illness as senile dementia. It took an editorial in the Archives of Neurology in 1976 by Robert Katzman to establish in the minds of researchers that these were the same disorder and to sound the alarm regarding its rising prevalence. The Alzheimer’s Association was established in the US a few years later in 1979.
That Alzheimer’s disease is not the only form of senile dementia has been realized still more recently with the emergence of frontotemporal lobar dementia (FTLD) as the second most prominent form of dementia in the young after Alzheimer’s disease. In Alzheimer’s disease, cognitive functions decline, beginning with learning and episodic memory (current and past experiences) and progressing to thinking, loss of language, and further decline in memory. In FTLD/FTD the first, clinically relevant signs involve changes in behavior and personality. As depicted in Fig. 1.6, each class of disease is associated with a specific region of the brain, each giving rise to distinct sets of clinical symptoms.
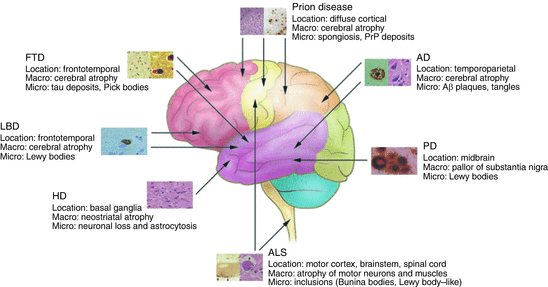
Fig. 1.6
Neurodegenerative disorders, their characteristics, and regions primarily affected. FTD frontotemporal dementia, LBD Lewy body disease, HD Huntington’s disease, ALS amyotrophic lateral sclerosis, PD Parkinson’s disease, AD Alzheimer’s disease (from Bertram J. Clin. Invest. 115: 1449 © 2005 American Society for Clinical Investigation)
One of the first sets of goals in the field was to characterize the extra- and intracellular deposits of proteins associated with the neurodegenerative diseases. As indicated in Fig. 1.6 extracellular and especially intracellular deposits are present not just in Alzheimer’s disease and Parkinson’s disease but in all the others as well. First, there was a need to identify the composition of the extracellular plaques and neurofibrillary tangles of Alzheimer’s disease, the Lewy bodies seen in Parkinson’s disease, and the others. Secondly, there was a need to tie the constituents whenever possible to genetic mutations in familial cases of the disease, or to other abnormalities, thereby providing evidence that these deposits are causally connected to the neurodegeneration. These efforts required the concerted application of an ensemble of newly developed biochemical and genetics techniques.
1.6.1 Modern Genetics Comes of Age
The modern field of genetics has its beginnings in the early 1900s with the efforts by the British geneticists William Bateson (1861–1926) and Reginald Punnett (1875–1967). Punnett introduced the public to the new field with his book Mendelism in 1905; Bateson coined the term “genetics” (taken from the Greek word for “to give birth”) in 1906, and the two of them launched the Journal of Genetics in 1910. Gregor Mendel (1822–1864) is well known today as the father of genetics. His laws of inheritance were neglected prior to their independent rediscovery in 1900 by Hugo de Vries and Carl Correns, and shortly thereafter Bateson and Punnett put forth the idea of gene linkage as an explanation of departures they observed from Mendel’s laws. With these activities the modern field of genetics was born. (The term gene linkage refers to the propensity of genes that are located in close proximity to one another on a chromosome to be passed on together during meiosis.)
These events were accompanied by the recognition that chromosomes were the seat of genes. The association of genes with chromosomes was first posited by Walter Sutton in 1903 and that identification was firmly solidified in 1915 by the publication of the studies by Thomas Morgan and his coworkers. The final step was working out the number of human chromosomes. That number was initially established in the early 1920s as 48, but was eventually and conclusively reduced to the correct number, 46, in 1956–1958. It is now well established that there are 22 paired somatic chromosomes; the 23rd, or sex chromosomes, consists of an XX pair for females and a XY pair for males. Each human chromosome has two arms—a short arm (denoted by the symbol “p”) and a long arm (indicated by the symbol “q”). These two arms are joined at the centromere.
1.6.2 Connections Are Established
The extracellular plaques seen in AD are composed of fibrils of the amyloid-beta protein, a proteolytically cleaved short peptide of around 40–42 amino acids generated from the genetically encoded amyloid precursor protein (APP). The first step in this identification came in 1984 when Glenner and Wong succeeded in determining the amino acid sequence for the principal protein found in the cerebrovascular amyloidosis associated with AD. They named this peptide the “amyloid-beta (Aβ) protein”. This same protein was isolated from extracellular plaque core by Masters in 1985.
Down syndrome is the leading cause of mental retardation, with a frequency of occurrence of about 1 per 700 births. It is named after the British physician John Langdon Down (1828–1896), who in 1866 published an essay in which he described the condition seen by him in the children’s asylum that he operated as superintendent. The association of Down syndrome with a genetic abnormality took nearly a century to establish. That this was indeed the case had been put forth in the 1930s but it was not until 1959 that Jérôme Lejeune (1926–1994) and Patricia Jacobs working independently identified its cause as the presence of an extra copy of chromosome 21, that is, there were three and not two copies of chromosome 21 (trisomy 21).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


