Lamotrigine
Frank G. Gilliam
Barry E. Gidal
CHEMISTRY AND MECHANISM OF ACTION
Lamotrigine is a phenyltriazine, a tertiary amine derivative [3,5-diamino-6-(2,3-dichlorophenyl)-1,2,4-trizine; molecular weight, 256.09] that is poorly soluble in water or alcohol (Fig. 60.1). Lamotrigine acts through voltage- and usedependent blockade of neuronal sodium channels, with greater blockade during repetitive activation (1, 2, 3, 4). Blockade of sodium channels activated from depolarized membrane potentials occurs at lower concentrations than those required to elicit blockade from hyperpolarized membrane but at clinically achievable concentrations (5). Lamotrigine appears to stabilize the inactivated state of the Na+ channel. Recently, a potential binding site within the Na+ channel pore was identified (6). In addition, lamotrigine dosedependently inhibits high-voltage activation of Ca2+ currents, possibly through inhibition of presynaptic N and P/Q-type Ca2+ channels (3,7, 8, 9). Despite apparent activity in human absence seizures, lamotrigine does not appear to inhibit low-voltage currents mediated by T-type Ca2+ channels. Although these actions are mechanistically similar to those of phenytoin, important differences do exist between these agents. Phenytoin inhibits veratrine-evoked release of glutamate and γ-aminobutyric acid (GABA). At similar concentrations, lamotrigine is twice as effective in inhibiting the release of glutamate compared to the release of GABA (10). Release of excitatory amino-acid neurotransmitters such as glutamate and aspartate are blocked during sustained repetitive firing. Animal models also suggest that lamotrigine inhibits ischemia-induced release of excitatory neurotransmitters (11, 12, 13, 14). Inhibition of nitric oxide release (15) and serotonin uptake (16) may also modestly contribute to lamotrigine’s action in both epilepsy and affective disorders. Lamotrigine appears to only modestly inhibit potassium channels and is a weak inhibitor of 5-HT uptake in humans or rodents (16). Lamotrigine is not an N-methyl-D-aspartate (NMDA) receptor antagonist (17) and does not appear to alter either plasma or brain GABA concentrations in humans (18,19).
Most likely, its antiepileptic actions and clinical spectrum can be explained largely by the combination of both Na+ and Ca2+ (N, P/Q) channel inhibition.
Lamotrigine prevents maximal electroshock seizures in mice, with potency and duration similar to those of phenytoin and carbamazepine, but does not prevent pentylenete-trazole-induced clonus, a model of absence seizures (20). Photically evoked afterdischarges and photoconvulsive responses are suppressed (21). Activity has been demonstrated in the genetic epilepsy-prone rat (22) and in the electrically induced electroencephalogram after-discharge model (23). Lamotrigine does not prevent cortical kindling in rats, but it does attenuate kindled seizures in a dose-dependent manner (23, 24, 25).
ABSORPTION, DISTRIBUTION, AND METABOLISM
Lamotrigine is an orally administered drug, available in various dosage strengths and bioequivalent formulations, including dispersible tablets. Lamotrigine is completely absorbed, with a bioavailability of 98% (26). Peak serum concentrations are achieved within 1 to 3 hours after oral administration (27). Lamotrigine displays linear oral absorption, with proportionality observed after doses up to 700 mg (28,29). A secondary peak in serum concentration between 4 and 6 hours after oral or parenteral administration suggests enterohepatic recycling. Food does not significantly affect absorption (30). Systemic absorption also occurs after rectal administration, although mean areas under the curve (AUC) are approximately 50% of corresponding oral values (31).
Approximately 56% of lamotrigine is bound to plasma proteins, and moderate binding remains constant over a concentration range of 1 to 10 μg/mL (28). In vitro, protein
binding is unaffected by phenytoin, phenobarbital, carbamazepine, or valproate (28).
binding is unaffected by phenytoin, phenobarbital, carbamazepine, or valproate (28).
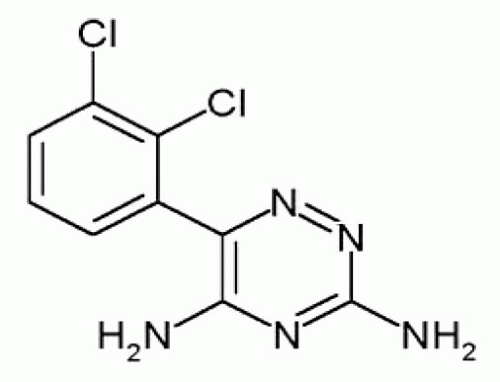 Figure 60.1 Structure of lamotrigine. |
Volume of distribution is independent of dose and ranges between 0.9 and 1.2 L/kg in healthy volunteers (28,30). Data from rodents and human ex vivo placental perfusion studies suggest that lamotrigine easily and rapidly crosses the placenta (32) and is present in maternal milk at potentially clinically significant levels (33). In humans, lamotrigine undergoes extensive hepatic metabolism by uridine diphosphate (UDP)-glucuronosyl-transferase (UGT 1A4) (34). Glucuronide conjugation can occur at both heterocyclic nitrogen atoms to form a quaternary amine glucuronide (35). In healthy volunteers, 70% of a single dose is recovered in the urine (28), with the 5-N and 2-N-glucuronide metabolite accounting for 90% of the recovered dose. This glucuronide metabolite is pharmacologically inactive. Renal elimination of unchanged drug represents less than 10% of an administered dose.
The elimination half-life of lamotrigine monotherapy in adults is approximately 24 to 29 hours. Oral clearance averages 0.35 to 0.59 mL/kg per minute (28,36). Clearance is higher in children and lower in the elderly than in young adults. The concentration to dose ratio was approximately 30% to 50% lower in 3- to 6-year-old children than in 7- to 15-year-old children or young adults (37). In 12 children between 4 and 11 years of age receiving monotherapy, mean oral clearance was 0.64 mL/kg per minute and elimination half-life was 32 hours (38). In a group of elderly volunteers between 65 and 76 years of age, clearance was 37% lower than in a group of 26- to 38-year-old adults (27).
Evidence suggests that lamotrigine undergoes autoinduction. Population analysis of sparse data obtained retrospectively from 163 patients receiving monotherapy demonstrated a 17% increase in clearance over 48 weeks (39). This modest degree of autoinduction is not clinically meaningful, however, because initiation of lamotrigine therapy usually involves gradual dose escalation.
Severity of hepatic disease can influence lamotrigine pharmacokinetics, and patients with Child-Pugh scores of 5 to 6 (B) or 7 to 9 (C) require respective dosage reductions of 50% to 75% (40). No significant differences in plasma clearance have been noted in patients with chronic renal failure (41). Approximately 17% of a lamotrigine dose may be removed by hemodialysis, with a corresponding reduction in half-life to about 13 hours (42).
The apparent oral clearance of lamotrigine does not appear to differ significantly between men and women (39) but may increase more than 65% during pregnancy, most noticeably during the second and third trimesters. Clearance returns to prepregnancy levels postpartum (43).
A serum concentration-effect range for lamotrigine has not been defined (44,45), and patients may respond to a wide range of drug concentrations. A target range between 4 and 14 μg/mL has been suggested for patients with epilepsy (46,47). Use of serum concentration data may be used to interpret drug interactions and compliance issues.
DRUG INTERACTIONS
Effect of Other Drugs on Lamotrigine
Comedication with Inducing Antiepileptic Drugs
Substantial interpatient variability in plasma clearance of lamotrigine can be explained largely by the presence or absence of concomitant drug therapy (48,49). Lamotrigine elimination half-life is reduced approximately 50% (about 12 to 15 hours) in the presence of UGT-inducing drugs, such as carbamazepine, phenobarbital, primidone, and phenytoin (50). While the addition of an enzyme inducer to a lamotrigine-containing regimen is well established, the time course of de-induction, after the removal of a concomitant inducer such as phenytoin or carbamazepine, is less clear-cut. An analysis of data derived from a pivotal conversion to monotherapy trial (51) found that mean lamotrigine plasma concentrations approximately doubled after the withdrawal of concomitant phenytoin; increases of only 50% to 75% followed the withdrawal of carbamazepine cotherapy (52). These data suggested that lamotrigine concentrations did not significantly change (increase) until concentrations of phenytoin or carbamazepine approached zero (52). Similar observations came as well from a small, prospective study (53).
Lamotrigine does not interact significantly with newer antiepileptic drugs such as gabapentin, zonisamide, levetiracetam, or felbamate, although its serum concentrations have been reported to be modestly decreased when concomitantly administered with either oxcarbazepine (54, 55, 56, 57) and may be significantly changed by felbamate (55) or topiramate (58). The clinical significance of these interactions is uncertain.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


