Lennox-Gastaut Syndrome
The definition of the Lennox-Gastaut syndrome (LGS) remains debated. The syndrome was originally described in 1966 by H. Gastaut, who proposed the use of the term Lennox syndrome to describe a childhood epileptic encephalopathy with diffuse spike-wave complexes and multiple types of attacks, including tonic seizures. The eponym was a tribute to the early work of Lennox and Davis (1950), who had described, in detail, the symptoms associated with slow spike-wave (SSW) activity in the electroencephalogram (EEG). The name Lennox-Gastaut syndrome was later adopted (Niedermeyer, 1969), and its use has gained wide acceptance even though the criteria for definition have been modified.
The term is often loosely used to denote severe epilepsy syndromes of childhood featuring several types of seizures, including falls, that are often medically intractable. Such a broad definition, however, encompasses several types of epilepsy for which the outcome and therapy differ. Recently, the tendency has been to limit the term LGS to a more narrowly defined syndrome from both the point of view of the seizures themselves and of the EEG characteristics (see Chapters 5 and 6).
Currently, LGS is defined as an epilepsy syndrome that is characterized by multiple types of seizures, including a nucleus of brief tonic or atonic seizures; atypical absences; and less characteristically, myoclonic attacks associated with an interictal EEG pattern of diffuse, slow (less than 2.5 Hz) spike-wave complexes (Commission on Classification and Terminology of the International League Against Epilepsy [ILAE], 1989). Several authors (Genton and Dravet, 1998; Beaumanoir, 1985; Beaumanoir et al., 1974) consider the presence of fast (10 Hz) rhythms associated with the tonic attacks or occurring with minimal or without associated clinical manifestations, especially during non-rapid eye movement (non-REM) sleep, an additional necessary criterion. Episodes of nonconvulsive status epilepticus are a common occurrence. Mental retardation is a highly common, but not an absolutely constant, feature (Aicardi and Gomes, 1991).
Even with this stricter definition, some disagreement still exists over the degree of subdivision that is desirable and the limits of accepted subsyndromes, and a fair proportion of cases resist precise classification because they display features of both LGS and myoclonic epilepsy; in some cases, the differentiation may rest on the quantitative proportions of the various types of seizures rather than on the specific features.
In addition to the suggestive types of seizures, other types of seizures and EEG features can occur with LGS; for example, focal seizures are often present either at the same time as or before the more typical types of attacks.
None of the aforementioned features is found in every case. Tonic seizures were present in 80% to 92% of patients (Gastaut et al., 1973a), and fast rhythms were seen during slow sleep in only 55% of those patients who had whole-night sleep recordings (Baldy-Moulinier et al., 1988). Moreover the “typical” seizures of LGS may have, in some patients, been preceded for prolonged periods by other types of attacks. This is especially true for myoclonic seizures, which may long be the prominent feature, leading some investigators to describe a “secondary type” of LGS that follows a phase of essentially myoclonic astatic epilepsy (Hoffmann-Riem et al., 2000).
As a result of these nosologic uncertainties, the features of LGS are variably described, depending on the definition that is accepted. In this chapter, the authors use the term LGS in the restrictive sense that is now accepted by most investigators (Beaumanoir and Dravet, 2003; Glauser and Morita, 2000; Genton and Dravet, 1998). The LGS syndrome is usually considered a secondary generalized epilepsy.
INCIDENCE AND ETIOLOGY
The frequency of LGS has been estimated as being between 1% and 10% of childhood epilepsies (Luna et al., 1988; Trevathan et al., 1997). The former figure is probably closer to reality, but a high incidence is observed in tertiary epilepsy centers that deal with intractable patients. The actual population incidence is poorly known because of the selection of patients for referral and the different definitions that are in use. Epidemiologic studies show that the proportion of LGS seems relatively consistent across various
populations. The prevalence of LGS in mentally retarded children was reported as being 0.06 per 1,000 (Steffenburg et al., 1998), and the percentage of LGS in institutionalized patients with mental retardation may be as high as 16.3% (Mariani et al., 1993). Male children are affected more often than female children (54% to 63%). In the vast majority, onset is between 1 and 7 years of age. The peak age at onset is between 3 and 5 years. Up to 20% of cases have their onset before the age of 2 years. However, cases with onset in late childhood and adolescence or even adulthood are known to occur (Roger et al., 1987).
populations. The prevalence of LGS in mentally retarded children was reported as being 0.06 per 1,000 (Steffenburg et al., 1998), and the percentage of LGS in institutionalized patients with mental retardation may be as high as 16.3% (Mariani et al., 1993). Male children are affected more often than female children (54% to 63%). In the vast majority, onset is between 1 and 7 years of age. The peak age at onset is between 3 and 5 years. Up to 20% of cases have their onset before the age of 2 years. However, cases with onset in late childhood and adolescence or even adulthood are known to occur (Roger et al., 1987).
LGS can be preceded by other forms of seizures, especially infantile spasms (Beaumanoir and Dravet, 1992; Ohtahara and Yamatogi, 1990; Hodge et al., 1989; Chevrie and Aicardi, 1972), and timing the transition from brief “clonic” spasms to the longer tonic seizure of LGS precisely is difficult. Other types of epileptic seizures preceding the appearance of LGS include unilateral seizures, generalized tonic-clonic seizures, and episodes of convulsive status epilepticus. Focal seizures are relatively common (Aicardi, 1991c; Aicardi and Gomes, 1991; Gastaut and Zifkin, 1988; Gastaut et al., 1973a, 1974b). Primary generalized seizures, including typical absences, have been observed only rarely (Beaumanoir and Dravet, 1992; Roger et al., 1989). Some cases of myoclonic absences may evolve to LGS (Tassinari et al., 1992b). Focal and generalized seizures may also precede the appearance of the syndrome.
One-fourth to one-third of cases occur in children without previous developmental or neurologic abnormalities and without evidence of brain damage on imaging. In the 1989 Classification of Epilepsies and Epileptic Syndromes (Commission on Classification and Terminology of the ILAE, 1989), such cases were termed cryptogenic because of the usual presence of mental retardation. However, this mental retardation may not be the consequence of a lesion but rather the result of the epileptic activity itself, so the use of the term idiopathic epilepsy, which is sometimes encountered (Ohtahara, 1988; Boniver et al., 1987), may be acceptable even though LGS is classically considered a symptomatic or cryptogenic epilepsy.
The etiology of LGS is heterogeneous. Brain damage plays a major role, whereas genetic factors are generally regarded as less important. However, the frequency of a family history of epilepsy varies considerably from 2.5% to 47.8% (Dravet and Roger, 1988). This discrepancy is probably due to the different diagnostic criteria used, and it reflects the nosologic problems raised by the syndrome.
Two-thirds to three-fourths of cases result from a demonstrable brain abnormality or occur in patients with previous developmental delay (Ohtahara, 1988; Chevrie and Aicardi, 1972), and these are termed symptomatic or secondary.
Most lesions responsible for LGS are of developmental origin. Abnormalities of cortical development were the cause of 10 of 30 autopsy cases reported by Roger and Gambarelli-Dubois (1988). Multiple types of cortical malformations have been found, including bilateral perisylvian and central dysplasia (Kuzniecky et al., 1993a; Guerrini et al., 1992c, 1992d; Ricci et al., 1992), diffuse subcortical laminar heterotopias (Palmini et al., 1991a), and focal cortical dysplasias (Palmini et al., 1991c; Guerrini et al., 1996a). A few cases are due to Sturge-Weber syndrome (Chevrie et al., 1988; Roger and Gambarelli-Dubois, 1988) or tumors, especially of the frontal lobes (Angelini et al., 1979). LGS has also been reported in cases of hypothalamic hamartomas following a long period of focal gelastic seizures (Berkovic et al., 1988). The pathologic basis of cases of LGS not due to macroscopic lesions is poorly known. Biopsy studies (Renier, 1988; Renier et al., 1988b) have shown only relatively minor changes, such as poor dendritic arborizations and disturbed synaptic development of pyramidal cells in the inner layers of the cortex.
Major brain malformations are less commonly a cause of LGS than they are of infantile spasms (Aicardi, 1986a, 1994). LGS has not been reported in Aicardi syndrome or with the lissencephaly syndrome, which suggests that gross damage does not permit the organization of rhythmic discharges.
Acquired destructive lesions are also less common. Hypoxic brain damage has been reported (Ohtahara, 1988; Roger and Gambarelli-Dubois, 1988), and large porencephalic defects can also produce a picture of LGS (Ohtahara, 1988; Palm et al., 1988).
LGS can follow other epilepsy syndromes. The most common preceding syndrome is West syndrome, which was found prior to the onset of LGS in 17.5% to 41% of cases (Donat, 1992; Aicardi and Gomes, 1991; Ohtahara et al., 1988). Cases following infantile spasms may represent a special subgroup of LGS, with an early onset, a predominance of tonic seizures occurring in clusters, and a particularly poor prognosis (Aicardi and Gomes, 1992; Donat and Wright, 1991a; Ohtahara, 1988).
Cases of LGS following myoclonic astatic epilepsy (Hoffmann-Riem et al., 2000) are not included by all investigators. Many authors state that, although the late course of myoclonic astatic epilepsy is often marked by the appearance of nocturnal
tonic seizures, usually the latter are not a prominent feature and they do not warrant the diagnosis of LGS. However, prominent tonic seizures may emerge after an initial history of myoclonic astatic seizures, and such cases are regarded by some investigators to fulfill the criteria for LGS because the differences, if any, are mainly quantitative and therefore are rather subjective. Such cases illustrate the difficulties in strictly delimitating LGS and differentiating it from some cases of related myoclonic epilepsy. Indeed, similar cases have been reported under the term myoclonic variant of the LGS (Dravet et al., 1982; Aicardi and Gomes, 1992).
tonic seizures, usually the latter are not a prominent feature and they do not warrant the diagnosis of LGS. However, prominent tonic seizures may emerge after an initial history of myoclonic astatic seizures, and such cases are regarded by some investigators to fulfill the criteria for LGS because the differences, if any, are mainly quantitative and therefore are rather subjective. Such cases illustrate the difficulties in strictly delimitating LGS and differentiating it from some cases of related myoclonic epilepsy. Indeed, similar cases have been reported under the term myoclonic variant of the LGS (Dravet et al., 1982; Aicardi and Gomes, 1992).
CLINICAL AND ELECTROENCEPHALOGRAPHIC ICTAL FEATURES
Seizures
The seizures of LGS are usually repeated, occurring many times daily. They include a nucleus of “core” seizures, which are mainly atonic, tonic, and atypical absence seizures. They are often associated with other less characteristic types as well. They are particularly frequent during sleep. However, the frequency of seizures can change considerably, with the individual having both “bad” and “good” periods.
Tonic Seizures
Tonic seizures are the most characteristic type (Roger et al., 1989; Chevrie and Aicardi, 1972), although various estimates have placed their frequency from 17% of the cases (Niedermeyer, 1969) to 55% (Chevrie and Aicardi, 1972) and up to 95% (Gastaut et al., 1973a). The higher incidence has been found in series in which sleep tracings were systematically obtained. Tonic seizures occur frequently during non-REM sleep and less frequently during wakefulness, and they do not occur during REM sleep. They are usually brief, lasting from a few seconds to 1 minute, with an average duration of about 10 seconds. Consciousness is lost or obscured during the seizures, although arousal from light stages of sleep may occur (Erba and Cavazzuti, 1981). The axial subtype consists of a brief, but sustained, bilateral symmetric contraction of the axial muscles that results in a flexor movement of the head and trunk with apnea that occasionally is preceded by a brief cry. The eyes open and often deviate upwards. Clouding of consciousness and autonomic manifestations are usually associated with this subtype. In axorhizomelic seizures, associated abduction and elevation of the arms occurs, whereas global tonic attacks involve most muscles and affect the distal parts of the limbs. When the child is standing, the flexion of the lower limbs and body axis may forcefully throw the patient to the ground. Patients who fall in a rigid posture, like a statue, are having a tonic seizure, whereas those who fall by collapsing may have either an atonic seizure or a global tonic seizure with triple flexion of the lower extremities (Erba and Browne, 1983). In all of the types, autonomic phenomena, including tachycardia, cyanosis, flushing of the face, salivation, and lacrimation, are common. Generally, eyelid retraction, staring, mydriasis, and cyanosis can be intense. Tonic seizures may be quite mild, especially in sleep, when they are often limited to a brief apnea, eye opening, an upwards deviation of the eyes, or a minimal stiffening, leading them to be easily mistaken for physiologic phenomena such as yawning or stretching. Axial spasms are very brief tonic seizures lasting 2 to 4 seconds that are a common cause of falls in patients with LGS (Egli et al., 1985; Ikeno et al., 1985). In infants, tonic seizures may tend to occur in clusters and to be followed by atonia, possibly making them difficult to distinguish from infantile spasms (Donat and Wright, 1991a; Roger et al., 1979). In older patients, episodes of automatic behavior may follow the tonic phase or may alternate with tonic contractions (Oller-Daurella, 1970). Such tonic automatic seizures have been found in 16% of patients (Roger et al., 1989).
The ictal EEG of tonic seizures may show a very fast discharge at a rhythm of 20 plus or minus 5 Hz that is usually of increasing amplitude or a discharge of 10 Hz with a high amplitude from onset; otherwise, it is similar to the “epileptic recruiting rhythm” (Brenner and Atkinson, 1982; Fariello et al., 1974; Gastaut et al., 1963, 1974a), lasting from 4 to 10 seconds in most cases. However, it may last from 30 to 60 seconds. Less commonly, a simple flattening of the tracing throughout the attack can be the only EEG manifestations (Fig. 4.1); the fast rhythm is followed by a brief discharge of spike-wave complexes or a burst of slow waves. The electromyographic (EMG) reading during tonic seizures resembles that of a normal tonic contraction.
Atypical Absence Seizures
Atypical absence seizures, which are observed in 13% to 100% of patients, are the second most common type of seizure in LGS (Aicardi and Gomes, 1992;
Gastaut et al., 1973a). They may closely resemble typical absences, although their onset and termination are said to be less abrupt or mild and they may be hardly detectable clinically because the loss of consciousness can be incomplete, allowing the child to continue his or her activities, albeit at a slower pace and imperfectly. The decreased consciousness is often associated with some loss of muscle tone, erratic myoclonic jerks, sialorrhea, or mild hypertonia of neck and back muscles. Relatively frequently, consciousness seems to be preserved. However, the execution of simple repetitive tasks is interfered with by the occurrence of the EEG discharge (Gastaut et al., 1974b; Porter et al., 1973; Goode et al., 1970). In some cases (Erba and Cavazzuti, 1977), a selective impairment of higher cortical functions in which responsiveness is maintained has been documented. The ictal EEG pattern of atypical absences is variable. The classic 3-Hz spike-waves are never observed (Beaumanoir and Dravet, 1992; Ohtahara, 1988; Porter and Penry, 1983; Goode et al., 1970), and this is often the only clear difference from typical absences. Often, a 10-Hz discharge that is similar to that recorded during tonic seizures is seen; in other cases, a burst of spike-waves, usually at 2.5 Hz or less, is recorded and is sometimes initiated by a brief phase of fast rhythm. A sudden voltage attenuation, sometimes with superimposed 20-Hz low-amplitude activity, may be present. Atypical absences are not precipitated by hyperventilation or photic stimulation. The duration of these discharges usually is 5 to 30 seconds. Identical discharges can be unassociated with clinical manifestations. The same applies to the discharges of fast rhythms that occur during slow sleep, although, in both situations, excluding the presence of minimal clinical phenomena can be difficult (Ohtsuka et al., 1982; Gastaut et al., 1963, 1974b).
Gastaut et al., 1973a). They may closely resemble typical absences, although their onset and termination are said to be less abrupt or mild and they may be hardly detectable clinically because the loss of consciousness can be incomplete, allowing the child to continue his or her activities, albeit at a slower pace and imperfectly. The decreased consciousness is often associated with some loss of muscle tone, erratic myoclonic jerks, sialorrhea, or mild hypertonia of neck and back muscles. Relatively frequently, consciousness seems to be preserved. However, the execution of simple repetitive tasks is interfered with by the occurrence of the EEG discharge (Gastaut et al., 1974b; Porter et al., 1973; Goode et al., 1970). In some cases (Erba and Cavazzuti, 1977), a selective impairment of higher cortical functions in which responsiveness is maintained has been documented. The ictal EEG pattern of atypical absences is variable. The classic 3-Hz spike-waves are never observed (Beaumanoir and Dravet, 1992; Ohtahara, 1988; Porter and Penry, 1983; Goode et al., 1970), and this is often the only clear difference from typical absences. Often, a 10-Hz discharge that is similar to that recorded during tonic seizures is seen; in other cases, a burst of spike-waves, usually at 2.5 Hz or less, is recorded and is sometimes initiated by a brief phase of fast rhythm. A sudden voltage attenuation, sometimes with superimposed 20-Hz low-amplitude activity, may be present. Atypical absences are not precipitated by hyperventilation or photic stimulation. The duration of these discharges usually is 5 to 30 seconds. Identical discharges can be unassociated with clinical manifestations. The same applies to the discharges of fast rhythms that occur during slow sleep, although, in both situations, excluding the presence of minimal clinical phenomena can be difficult (Ohtsuka et al., 1982; Gastaut et al., 1963, 1974b).
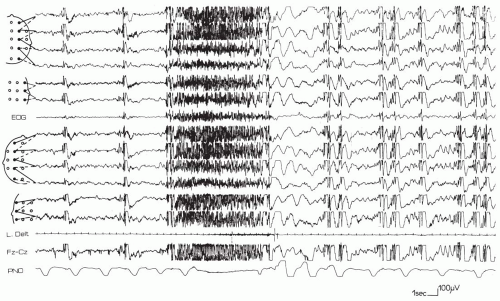 FIG. 4.1. A 12-year-old girl with Lennox-Gastaut syndrome experiences a tonic seizure during recording while asleep. A discharge of high-amplitude fast rhythms lasting about 10 seconds is accompanied by a slightly progressive tonic contraction that is apparent on the recorded muscle (left deltoid), and it is accompanied by apnea. The end of the seizure is characterized electrically by a discharge of polyspike and spike-wave complexes and a suppression burst and clinically by a short series of slow waves, followed by bursts of generalized multiple spike-wave complexes. Abbreviation: PNO, pneumogram. |
Myoclonic Seizures
Myoclonic seizures are considered less characteristic and less common in LGS than tonic seizures or atypical absences. Their frequency has been variably
estimated at 11% to 28% of cases (Beaumanoir and Dravet, 1992; Geoffroy et al., 1983; Dravet et al., 1982; Gastaut et al., 1973a; Chevrie and Aicardi, 1972). Myoclonic attacks are extremely brief, shocklike muscle contractions that may be isolated or repeated in a saccadic manner, usually for only a few seconds. The jerks are often bilateral and symmetric (massive myoclonus), and they preferentially involve the axial flexor muscles and the abductors of both arms. The involvement of the lower limbs can cause the patient to fall to the ground. No appreciable loss of consciousness occurs. However, myoclonias may be quite prominent in some patients with LGS (the so-called myoclonic variant). They are probably a common cause of the falls that constitute a major problem for children with LGS, although the mechanism of the fall may be impossible to determine without polygraphic recording. The EEG concomitants of most myoclonic seizures are bursts of generalized polyspike-waves that often have a frontal predominance. Recently, in a small group of patients, Bonanni et al. (2002) showed that the myoclonus in LGS is not really bilateral and synchronous, like that of idiopathic myoclonic epilepsies, but that it was constantly generated in one hemisphere with rapid generalization consistent with callosal transmission, which suggests that the origin of LGS may be focal in at least a proportion of the cases.
estimated at 11% to 28% of cases (Beaumanoir and Dravet, 1992; Geoffroy et al., 1983; Dravet et al., 1982; Gastaut et al., 1973a; Chevrie and Aicardi, 1972). Myoclonic attacks are extremely brief, shocklike muscle contractions that may be isolated or repeated in a saccadic manner, usually for only a few seconds. The jerks are often bilateral and symmetric (massive myoclonus), and they preferentially involve the axial flexor muscles and the abductors of both arms. The involvement of the lower limbs can cause the patient to fall to the ground. No appreciable loss of consciousness occurs. However, myoclonias may be quite prominent in some patients with LGS (the so-called myoclonic variant). They are probably a common cause of the falls that constitute a major problem for children with LGS, although the mechanism of the fall may be impossible to determine without polygraphic recording. The EEG concomitants of most myoclonic seizures are bursts of generalized polyspike-waves that often have a frontal predominance. Recently, in a small group of patients, Bonanni et al. (2002) showed that the myoclonus in LGS is not really bilateral and synchronous, like that of idiopathic myoclonic epilepsies, but that it was constantly generated in one hemisphere with rapid generalization consistent with callosal transmission, which suggests that the origin of LGS may be focal in at least a proportion of the cases.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


