Less Common Epilepsy Syndromes and Epilepsy Associated with Chromosomal Abnormalities
Sophia Varadkar
J. Helen Cross
The past 10 years have seen a dramatic increase in the number of described epilepsy syndromes, as evidenced by the proposed revisiting of the International League Against Epilepsy (ILAE) classification (2001). Debate over the revised classification continues, centering, in particular, on when a new syndrome should be “accepted” as opposed to “proposed.” Several epilepsy syndromes are described on the basis of specific clinical, neurophysiologic, and neuroimaging findings, and some are not included in the ILAE classification (2001). This chapter discusses rare but well-defined epilepsy syndromes, mainly those of early infancy, but also those associated with chromosomal abnormalities and hemiconvulsion-hemiplegia-epilepsy (HHE).
EPILEPSIES IN INFANCY
Epilepsy in infancy remains a challenge, both in diagnosis (symptomatic as opposed to idiopathic syndromes) and in treatment. In general, epilepsy during the first year of life is widely believed to carry a poor prognosis for seizure control and developmental outcome. However, a group of more benign epilepsies probably are underdiagnosed in view of their prompt response to medication and good developmental outcome.
Migrating partial seizures of infancy, early infantile epileptic encephalopathy (EIEE), and early myoclonic encephalopathy (EME) are aggressive, resistant syndromes that have a devastating impact on neurodevelopment. EIEE and EME are characterized by burst suppression on the electroencephalogram (EEG). Differentiating them from infantile spasms and other malignant epilepsies has therapeutic and prognostic implications. EIEE, in particular, has led to the concept of “age-dependent epileptic encephalopathy,” a disorder that evolves to infantile spasms and later to Lennox-Gastaut syndrome (1,2).
Benign myoclonic epilepsy of infancy (BMEI) and benign partial epilepsy of infancy share features with these aggressive syndromes but have a more favorable neurodevelopmental outcome and respond better to treatment. Both BMEI and severe myoclonic epilepsy of infancy (SMEI) may follow febrile convulsions but have a very different course. Distinguishing between them, however, may require initiation of treatment and observation over time.
Symptomatic Generalized Epilepsies
Early Myoclonic Encephalopathy
EME, like EIEE, begins neonatally and carries a poor prognosis (3). It affects both sexes equally, and familial cases have been reported. Because EME is associated with inherited metabolic disorders, such as (most commonly) nonketotic hyperglycinemia, propionic aciduria, methylmalonic
acidemia, D-glycine acidemia, and pyridoxine dependence (4,5), careful metabolic investigation of all cases is required.
acidemia, D-glycine acidemia, and pyridoxine dependence (4,5), careful metabolic investigation of all cases is required.
Seizures always begin before age 3 months, although overall onset is later than that in EIEE. Whereas the main seizure type in EIEE is tonic spasms, often erratic myoclonus (Fig. 31.1) and frequent partial seizures predominate in EME (6). Massive myoclonias and tonic spasms are sometimes seen, but erratic myoclonus is not a feature of EIEE. Evolution to infantile spasms has been reported with nonketotic hyperglycinemia (7).
The EEG shows burst suppression that is enhanced by sleep (6,8) and may not always be apparent in wakefulness. This pattern may persist but does not evolve with age.
Response to anticonvulsant medication is poor. Sodium valproate remains the first choice, and lamotrigine in combination may be helpful (9). Many children die by 2 years of age, and neurodevelopmental outcome in survivors is dismal. Continued deterioration to death is expected.
Early Infantile Epileptic Encephalopathy (Ohtahara Syndrome)
Still best known by its eponymous title, EIEE was first reported by Ohtahara in 1976 and is the earliest of the age-dependent epileptic encephalopathies. EIEE is characterized by early tonic seizures, a burst-suppression electroencephalographic pattern, poor response to treatment, and poor neurodevelopmental outcome, with evolution to infantile spasms and Lennox-Gastaut syndrome (1,10,11).
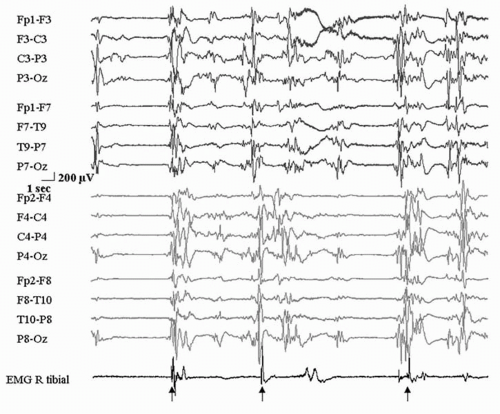 Figure 31.1 Early myoclonic encephalopathy. Electroencephalogram from a 3-month-old child with myoclonic jerking since before birth. Arrows indicate clinical jerks of the right leg. Note the markedly abnormal, high-amplitude, discontinuous electroencephalographic activities and the isolated myoclonias on the surface electromyographic recording. |
No cause is found in most cases, but many associations have been noted, most frequently structural brain anomalies. Aicardi syndrome, migrational disorders, porencephaly, and hemimegalencephaly are other accompanying conditions. Detailed neuroimaging is always warranted. There is no difference between male and female occurrence, and no familial cases have been reported.
Although seizures may begin on the first day of life, most start in the first month and nearly all by 3 months of age. Mainly tonic, the seizures occur in wakefulness and in sleep, singly or in clusters. Partial, focal motor, hemiconvulsion, and, rarely, myoclonic are other types. The typical burst-suppression pattern is seen during both waking and sleeping states and is periodic and consistent. Bursts of high-voltage slow waves and spikes last 1 to 3 seconds, followed by flattening for 3 to 4 seconds. Both the electroencephalographic pattern and the seizures show an age-dependent evolution: infantile spasms emerge clinically and hypsarrhythmia electrographically, either between 3 and 6 months or between 1 and 3 years of age, with later appearance of Lennox-Gastaut syndrome (12). Seizures are highly resistant to anticonvulsant therapy; corticotropin may produce a partial response and zonisamide is useful (13,14). Structural anomalies may be amenable to epilepsy surgery (15,16). Very poor developmental outcome and death in infancy or severe handicap are expected.
Migrating Partial Seizures of Infancy
This rare syndrome, beginning in the first 6 months of life, was first described by Coppola and coworkers in 1995, and
there are now more than 40 cases in the literature. The characteristic multifocal seizures show migration of the focus during the seizure. Cognitive regression, developmental devastation, and high mortality are expected outcomes (17,18).
there are now more than 40 cases in the literature. The characteristic multifocal seizures show migration of the focus during the seizure. Cognitive regression, developmental devastation, and high mortality are expected outcomes (17,18).
Males and females are equally affected and present at a mean age of 3 months with frequent partial seizures, initially with particular motor features (17,19). Autonomic manifestations may be striking; secondary generalization may be seen; and status epilepticus may occur (17,19). This initial period lasts an average of 6 to 7 weeks and is followed by months during which seizures increase in frequency, may occur in clusters, and vary in clinical expression. Developmental regression is dramatic (20); infants usually become progressively hypotonic and microcephalic and may demonstrate a movement disorder. The EEG shows nearly continuous seizures involving multiple independent areas. The focus may originate in either hemisphere and migrate from one area of the cortex to another during a single seizure and between consecutive seizures. This characteristic bilateral involvement is usually apparent from early in the disease. Its appearance up to 2 months into the illness may delay diagnosis (21). Interictally, multifocal epileptiform activity continues.
No risk factors have been recognized; a familial tendency seems lacking; extensive investigation has not identified a cause (18). In some cases, postmortem examination has shown hippocampal neuronal loss and gliosis (17).
Response to conventional antiepileptic drugs, steroids, a ketogenic diet, human immunoglobulin, and flunarizine has been disappointing (22). Therapy with oral potassium bromide achieved complete control in one infant and almost complete control in another; both infants evinced some developmental gains on short-term follow-up (23). Response to stiripentol and high doses of clonazepam has been inconsistent (24).
Benign Infantile Epilepsies
Benign partial epilepsy of infancy and BMEI are rare idiopathic syndromes whose diagnosis can be established only after treatment has begun and the benign course has been confirmed. Benign idiopathic and familial neonatal convulsions also have good prognoses but are not discussed here.
Benign Partial Epilepsy of Infancy
Partial epilepsies in infancy differ in etiology, therapy, and prognosis from the generalized epilepsies in this age group that may be manifested by focal seizures. The majority of focal epilepsies in infancy are symptomatic and are caused by neurocutaneous syndromes, brain malformations, focal dysplasias, and tumors. Described by Watanabe and colleagues in 1987 (25), benign partial epilepsy of infancy appears between 3 and 20 months of age. Radiologic and metabolic investigations are negative, although an increased family history of febrile and infantile convulsions may be present (25). Often occurring in clusters, the seizures are characterized by behavioral arrest and staring, with limb or facial automatisms and prompt secondary generalization in many (26). The EEG is focal at onset with recruiting rhythms. During the seizure, slow waves emerge and dissipate, and, finally, spikes, polyspikes, and sharp-wave complexes are evident in centrotemporal, occipital, and parietal regions bilaterally. The interictal EEG is unremarkable (27). Seizures are easily controlled with carbamazepine or phenobarbital but if untreated may infrequently occur in brief isolated clusters. In retrospect, treatment may not be necessary but is usually undertaken before the diagnosis becomes apparent. Development is normal prior to seizure onset and remains so (26). No association with later epilepsy has been identified. A familial syndrome has been described, with mean seizure onset at a slightly younger age, similar seizure semiology, excellent response to treatment, and normal development (28).
Benign Myoclonic Epilepsy of Infancy
In 1981, Dravet and Bureau described BMEI in infants and children between 4 months and 3 years of age. In contrast to early infantile myoclonic encephalopathy, BMEI involves a favorable neurodevelopmental outcome and usually easy seizure control (29, 30, 31). It accounts for approximately 1% to 2% of epilepsies beginning in infancy and early childhood. Development is normal; febrile convulsions may have occurred; 30% of infants have a family history of febrile convulsions or epilepsy (32). Clinical examination and neuroimaging are normal. More males than females are affected.
The myoclonic jerks—brief tonic contractions of the trunk and upper limbs and head drop—may be subtle and unrecognized at first; later they become more obvious and more intense, occurring in short series at any time of the day but more likely when the child is drowsy. Some seizures have an acoustic or tactile “reflex” trigger (33). Other seizure types are not expected.
The EEG shows normal background activity. Generalized spike-wave or polyspike-wave discharges, associated with the myoclonic jerks (32), are activated by drowsiness and during the first stages of sleep. Clinical and electroencephalographic photosensitivity are present in one-third of patients. The interictal EEG is normal for age.
Other conditions considered in the differential diagnoses must be ruled out. Infantile spasms and EME are distinguished by their terrible impact on development. SMEI and BMEI both demonstrate photosensitivity, but there is usually a history of status epilepticus and the later emergence of jerks in SMEI. The runs of myoclonic jerks in BMEI are short compared with those in infantile spasms; they appear early in BMEI. Other than febrile convulsions, no other seizure type is seen in BMEI, whereas multiple seizure types characterize SMEI,
Lennox-Gastaut syndrome, and myoclonic-astatic epilepsy. Development in BMEI continues normally despite the seizures; this is not the case in any of the other epilepsies just described.
Lennox-Gastaut syndrome, and myoclonic-astatic epilepsy. Development in BMEI continues normally despite the seizures; this is not the case in any of the other epilepsies just described.
Monotherapy with sodium valproate, the drug of choice, is generally efficacious. A good response to add-on therapy with phenobarbital and benzodiazepines has been reported, but ethosuximide and primidone have produced disappointing results. Therapy can usually be withdrawn by 6 years. Generalized tonic-clonic seizures have been observed between 9 and 12 years of age and on drug withdrawal (29,34,35). If withdrawal seizures occur or if photosensitivity persists, treatment continues.
With early diagnosis and institution of treatment, most infants will be seizure free and incur no educational or psychological problems. Despite this optimistic prognosis, however, and the “benign” designation, as many as 25% of patients in some series (35) experience mild learning difficulties.
EPILEPSIES ASSOCIATED WITH CHROMOSOMAL ANOMALIES
Epilepsy is increasingly recognized as a frequent and significant part of the clinical problems associated with chromosomal anomalies. As ongoing investigation documents the involvement of the central nervous system in many chromosomal abnormalities, more patients with epilepsy are noted to have chromosomal anomalies and more chromosomal anomalies are recognized to include epilepsy. Loss or abnormal function of genetic loci on involved chromosomes may lead to increased seizure susceptibility, cortical excitability, or changes in neurotransmitters. Alternatively, epilepsy may originate from structural brain abnormalities occurring with the chromosomal abnormality.
Although epilepsies in the setting of chromosomal anomalies range from very mild to severe, they are generally difficult to treat. In a few, such as Angelman syndrome, the epilepsy or the electroencephalographic pattern is characteristic or highly suggestive of the chromosomal anomaly; in the majority, this is not the case. A deepening knowledge of these epilepsies and their electroencephalographic patterns may contribute to their diagnosis, or, conversely, the chromosomal anomalies may suggest target genes for the investigation of seizure pathogenesis (36).
Ring Chromosomes
Epilepsy associated with ring chromosomes 14 and 20 is well recognized. Infantile spasms and hypoplasia of the corpus callosum have been reported with ring chromosome 9 (37). Epilepsy also has been noted with ring chromosomes 15, 17, and 21 (38, 39, 40).
Ring Chromosome 20
The incidence of ring chromosome 20 is unclear, and the syndrome is probably underdiagnosed, as the phenotype does not immediately suggest a chromosomal anomaly. The clinical features are mild to moderate learning difficulties, behavioral problems (usually restlessness and aggression), and epilepsy, often without significant dysmorphism. Usually present are atypical absences and nonconvulsive status with diffuse, slow, rhythmic electroencephalographic and less frequently perioral and eyelid myoclonia (41,42). Bifrontal slow waves can be a striking interictal feature (Fig. 31.2). Subtle nocturnal frontal lobe seizures also have been described (43). Adverse psychological events triggering episodes of status (44) may contribute to misdiagnosis of nonepileptic seizures.
Seizures often resist treatment, and no medication has been found either to be useful or to aggravate seizures. Excision of a cortical dysplasia in one patient did not resolve seizures (22). Vagus nerve stimulation succeeded in one patient (45). Interestingly, the chromosomal telomeric regions p13 and q13 lost in formation of the ring have been implicated in benign neonatal familial convulsions and in autosomal dominant nocturnal frontal lobe epilepsy, yet the actual candidate genes do not seem to have been lost in the cases examined.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


