Malformations of Cortical Development
Ruben I. Kuzniecky
Graeme D. Jackson
Introduction
The development of the human brain is a long and complex process that begins with the induction of the neural plate from the undifferentiated surface ectoderm and continues after birth.115 Any disruption of the normal mechanisms responsible for the formation of the cerebral structures can result in malformations due to abnormal cortical development.81,85 A wide variety of genetic and environmental factors can cause disturbances in these developmental processes and can therefore lead to an abnormality in the mature brain.
Until the advent of high-resolution magnetic resonance (MR), malformative disorders of the nervous system were almost exclusively the domain of the pathologist. With magnetic resonance imaging (MRI), abnormalities of cortical development can be identified in life, and understanding these conditions, their clinical consequences, and outcome has become essential for appropriate management. The physician’s goal is to diagnose these disorders accurately, using information that is available in the clinical setting. This chapter presents an approach that we believe is helpful to clinicians dealing with these disorders, especially in the setting of epilepsy.
The clinical circumstances in which these disorders are encountered are many but primarily involve developmental delay, epilepsy, skin lesions in specific neurocutaneous disorders, and associated organ malformations. From the perspective of epilepsy, there are two common presentations. First, a patient with epilepsy has an abnormal MR brain scan suggesting a malformation of cortical development (MCD). In this case, the issue is largely one of diagnosis, appropriate classification, knowledge of the relevant condition, and genetic testing, if available. This is important for prognosis, treatment, and genetic counseling. Second, a patient with epilepsy has a “normal” brain MR scan, but the clinician suspects, perhaps on the basis of family history, seizure intractability, or other findings on clinical examination, that there may be an underlying abnormality of cortical development. In this case, one is usually dealing with a subtle, localized cortical malformation, and the challenge is to identify the abnormal brain region(s). This is not an uncommon problem for epilepsy centers that deal with surgical treatment.
Clinical Presentation
Disorders of cortical development encompass many types of malformations with a comparably wide range of etiologies that produce different effects depending on the stage of brain development that is affected. Not surprisingly, then, clinical presentations are quite heterogeneous and can manifest at almost any age. As a result, the practical problem is that because there are actually many disorders of cortical development, there are no specific clinical features associated with MCDs when considered as a group.
While MCDs are a common cause of epilepsy, there are many cases in which seizures are not a feature of these disorders. Why almost identical brain abnormalities can have such variable clinical phenotypes is not known. Yet while there are no clinical features that are specific for MCDs taken as a whole, there are within this group some specific syndromes recognized on the basis of characteristic patterns of genetic, clinical, and imaging findings.
Seizure type usually reflects the topology of the malformations. That is, focal seizures occur with focal or multifocal MCD, and secondarily generalized seizures with diffuse or bilateral MCDs.
Almost any epilepsy presentation, at almost any age, can be due to an MCD. However, in focal epilepsy, some features create a strong suspicion of an underlying MCD and encourage thorough investigation to exclude this possibility if no other cause has been identified. These features include developmental delay, static focal neurologic deficits, a family history of developmental delay or epilepsy, frequent seizures from onset, and focal status epilepticus. While the presence of such elements may raise the suspicion of an MCD, it must be emphasized that none of them is specific. In surgical series of patients, such characteristics will likely lead to detailed high-resolution MRI with special techniques in an effort to define an abnormality of cortical development.
The severity of seizures in patients with MCDs also varies greatly. There are many individuals with extensive cortical malformations who have no seizures. On the other hand, some individuals with apparently small developmental malformations have severe and intractable epilepsy. The mechanisms of epileptogenesis associated with these abnormalities are complex and generally poorly understood.
Epidemiology
Few studies have addressed the epidemiology of MCDs in detail, and little information can be obtained from studies using the modalities, including MRI, that are currently available. One series based on pathology findings revealed that 46.5% of patients had developmental malformations at autopsy.95 A case ascertainment study of lissencephaly showed a prevalence of 11.7 per million births.40 No data are available for heterotopia, focal cortical dysplasia, or other malformations of cortical development.
Recent clinical and neuroimaging studies in special populations have suggested that cortical malformations are much more common than was previously appreciated. In children referred to epilepsy centers for intractable seizures, more than half have some type of developmental abnormality, with focal cortical dysplasia discovered in approximately 25% of those with intractable focal seizures.79 The prevalence of these
disorders in the adult population with intractable focal epilepsy is 15% to 20%.6,10
disorders in the adult population with intractable focal epilepsy is 15% to 20%.6,10
Terms and Definitions
A number of terms are commonly used in referring to patients with developmental malformations.10 For example, dysplasia in this context is an encompassing term that usually means abnormalities of the cortex that have a particular histopathology and are developmental in origin. Neuronal migration disorder is generally used in a similar context, but this is clearly incorrect as a general term, as it describes only one embryologic stage in cortical development. We consider all of these disorders to be malformations of cortical development and prefer this as the general term for referring to this category of conditions. All of these disorders seem to result from disturbed organogenesis (and, hence, are malformations), and all involve cells that under normal circumstances would participate in formation of the cerebral cortex. The most common malformative disorders involve abnormal stem cell formation in the germinative zone or abnormal cortical organization. Some result from faulty neuronal migration, whereas still others are postmigratory in origin.
We believe that establishing a single nomenclature and classification system for these disorders is essential to their understanding and management. The classification proposed in the following discussion provides a means by which similar disorders can be logically grouped together.10
Classification Principles
MCDs can be classified according to a number of different criteria emphasizing clinical phenotype, imaging findings, pathology, and genetic defects. The overall classification scheme that we favor (Table 1) is based on the three fundamental events of cortical formation: (a) proliferation of neurons and glia in the ventricular zone and subventricular zones; (b) multidirectional migration of immature but postmitotic neurons to the developing cerebral cortex; and (c) cortical organization, which consists of vertical and horizontal organization of neurons within the cortex and elaboration of axonal and dendritic ramifications. For those malformations with abnormalities involving more than one of these processes, classification is based on the first identified abnormal step. Diffuse and focal malformations that were classified separately in the past are no longer separated since genetic studies have shown that the same gene defects can cause focal or generalized MCDs.
Table 1 Classification Scheme of Malformations of Cortical Development | |
|---|---|
|
Thus, with advances in molecular genetics, we have moved from a purely phenotypic approach to a combined phenotypic/genetic classification. The basis of this change has been the recognition that malformations of varying severity can result from the same underlying processes, specifically from mutations of the same causative genes. This was shown first for classical lissencephaly38,93,94,111,112,113,114,123 and more recently in the brain malformations associated with congenital muscular dystrophies.15,16,17,88,89,108,132,133,134 For example, patients with large deletions and truncations of the LIS1 and DCX mutations have diffuse or severe lissencephaly, while those with less severe LIS1 mutations may only have posterior pachygyria or posterior-predominant subcortical band heterotopia. Those with less severe DCX mutations have anterior pachygyria or frontal subcortical band heterotopia of variable thickness,27,49,50,51,87,92,111,114,123 or they may even have normal brain MRI scans.57 Further support for this approach has been the recent discovery of mutations of multiple genes each causing very similar clinical syndromes, and the finding that mutations of different genes can cause the phenotype of Walker-Warburg syndrome or muscle-eye-brain disease.123
Developmental Malformations Associated with Epilepsy: Specific Disorders
We have restricted the following discussion to an overview of the most common and relatively distinct entities that affect patients with epilepsy.
Focal Cortical Dysplasia
Focal cortical dysplasia (FCD) is probably the most common form of focal developmental disorder diagnosed in patients with intractable focal epilepsy.24,67,83,86,100 The lesions consist of disruption of cortical lamination with poorly differentiated glial cell elements. Since its original description, FCD has been recognized to encompass a spectrum of changes.130 These range from mild cortical disruption without apparent giant neurons to the most severe forms in which cortical dyslamination, large bizarre cells, and astrocytosis are present.75,79,130 It is the presence of balloon cells that differentiates FCD type I (without balloon cells) from FCD type II (with balloon cells) and that lead to the distinction in our classification scheme (see Table 1).
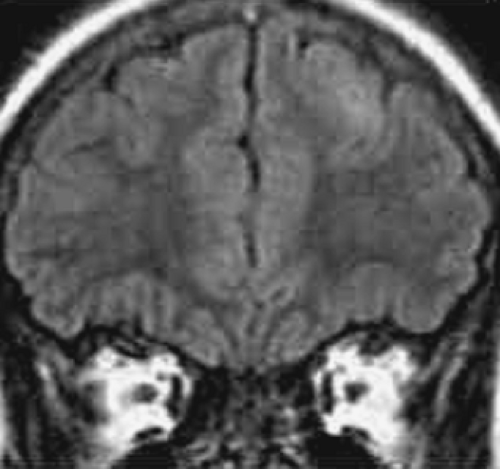 FIGURE 1. Focal cortical dysplasia (FCD). Left frontal lobe shows in this coronal fluid-attenuated inversion recovery magnetic resonance image a subtle signal abnormality and thickened cortex representing FCD. Pathology showed balloon cells. |
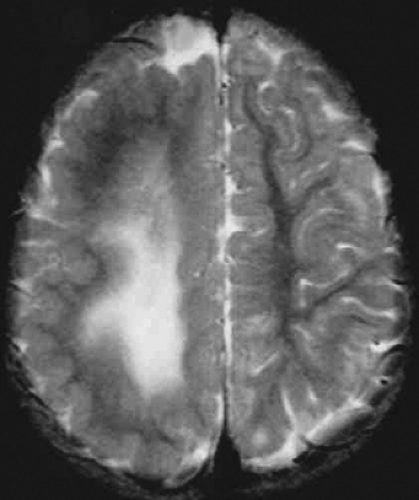 FIGURE 2. Hemimegalencephaly. Axial T2-weighted magnetic resonance image shows abnormal left hemisphere with smooth cortex and white matter changes. |
The clinical manifestations of patients with cortical FCD are variable. Seizures usually begin between the ages of 2 and 10 years. Sometimes, however, seizures may be the presenting clinical problem in the second decade or even later. Focal and secondarily generalized attacks are common. Interestingly, seizures often occur in clusters, but generalized status epilepticus is rare except in patients with FCD involving the central region.76 In our experience, the majority of patients have extratemporal cortical dysplasias that affect the pre- and postcentral regions most often. Interictal scalp electroencephalography (EEG) may demonstrate focal subclinical ictal discharges over the dysplastic lesions, underscoring the high epileptogenicity of these lesions.103 FCD involving the frontal lobe has also been reported, and lesions can occur in both mesial and lateral neocortical structures as well.77
The MRI findings consist of abnormal gyral thickening with underlying T2-weighted white matter changes. These abnormalities are often circumscribed in nature, and they can sometimes be extensive, involving more than one gyrus or lobe. High-resolution MRIs with thin slices and multiplanar reconstruction are often necessary to identify these24,25,55 (Fig. 1). Location can be quite unpredictable, as in the example of a very small lesion that was restricted to the bottom of a sulcus. Correlating clinical manifestations with the spectrum of changes seen in FCD has been limited, because histopathology is usually required before subtypes of FCD (e.g., FCD without balloon cells and microdysgenesis) can be firmly established.10
Hemimegalencephaly
Hemimegalencephaly is a rare malformation characterized by predominantly unilateral cerebral pathology typically associated with an enlarged hemisphere. It can be seen in isolation or in association with epidermal nevus syndrome105 or hypomelanosis of Ito.91,104,128 Pathologic findings are diverse and include cortical dysplasia, white matter abnormalities with abnormal cell types, or polymicrogyria usually restricted to one hemisphere. Most children with hemimegalencephaly associated with cortical malformations have not had other associated congenital anomalies.
Seizures and hemiparesis are common presenting symptoms.101,107,118 Developmental delay is also common. Seizures usually appear within the first 6 months of life. They are often unilateral but can secondarily generalize, and they are frequently intractable to medical therapy. Continuing seizure activity is associated with the appearance or worsening of unilateral neurologic findings, such as hemiparesis and hemianopias. Occasionally there is only minimal neurologic dysfunction.
Diagnosis is based on the predominantly focal epileptic syndrome, the unilateral hemispheric EEG discharges, and the presence of unilateral neurologic abnormalities. MRI findings provide definitive diagnosis. Mild to severe enlargement of at least one lobe is present in all patients (Fig. 2). In more than half of the patients, the entire hemisphere is enlarged with thick gray
matter and broad, flat gyri.23,83,136 The underlying hemispheric white matter usually demonstrates abnormal MRI signal intensity. Heterotopia and other malformations are sometimes detected throughout the abnormal hemisphere, and there may be ipsilateral ventricular enlargement.
matter and broad, flat gyri.23,83,136 The underlying hemispheric white matter usually demonstrates abnormal MRI signal intensity. Heterotopia and other malformations are sometimes detected throughout the abnormal hemisphere, and there may be ipsilateral ventricular enlargement.
Focal Transmantle Dysplasia
Transmantle dysplasia is a developmental malformation characterized by abnormal brain tissue that extends through the entire mantle of the cerebrum, from the pia to the ventricular surface.12 On MRI brain scans, areas of signal abnormality extend radially inward from the cortical surface toward the lateral ventricle. When the imaging plane is parallel to the tract, the T2 imaging signal abnormality involves the deep cortex and subcortical white matter.
Patients with this malformation present with focal seizures at different ages. Ictal semiology and neurologic deficits resemble those that accompany FCD. EEG abnormalities are usually lateralized to the areas of cortical malformations. Mild motor signs may be present if the lesions are in close proximity to the central cortical area. This malformation likely represents a subtype of FCD as pathology obtained during resections for epilepsy show a lack of normal cortical lamination, neuronomegaly, and hypomyelination with atypical reactive astrocytosis in the white matter.
Tuberous Sclerosis
Tuberous sclerosis (TS) is an autosomal dominant, genetically determined multisystem disorder with high penetrance and variable expression.35,36,37,71 Genetic heterogeneity is observed with gene defects reported in both chromosome 9q34 and chromosome 16p13 with genetic classification into TSC1 and TSC2.
Pathologically, TS is a disorder of cellular migration, proliferation, and differentiation, resulting in hamartomata formation that involves a large number of neural crest derivatives. Histologically, two major abnormalities are seen. Cortical tubers are characterized by cortical dyslamination, large cells (neurons and glial cells or neuroastrocytes), and abnormal neuropil with hypomyelination. Subcortically, subependymal nodules projecting into the ventricles are typical. Microscopically, densely aggregated larger cells are present, often resembling neoplasms. Electron microscopic (EM) studies of cortical tubers have demonstrated that glial cells predominate near the pial surface, whereas small neurons are more prevalent inferiorly. Underneath the tubers, a rudimentary cortical plate is seen. The presence of the most undifferentiated cell types in the subependymal zone (giant cells) and more differentiated cells in the cortical tubers with intermediate lesions between them suggest a spectrum of abnormalities in neural and glial differentiation and migration.
Seizures are the most common neurologic symptom in TS: More than 90% of patients have seizures during their lifetimes.71,120,142 Infantile spasms and partial seizures are highly prevalent, and secondary generalization occurs more often after age 2 years. Myoclonic seizures and mental retardation are very frequent. Over time, clinical deterioration is common, with seizures becoming more frequent and difficult to treat.
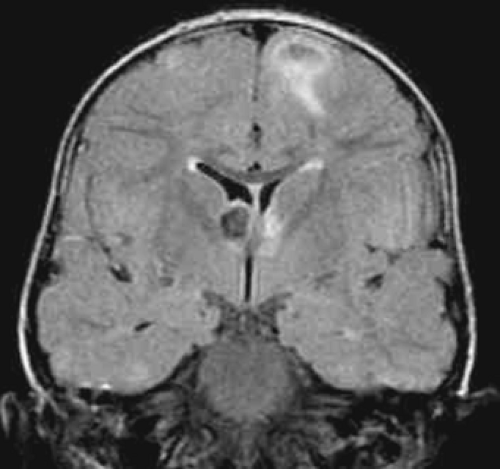 FIGURE 3. Tuberous sclerosis complex. Coronal fluid-attenuated inversion recovery magnetic resonance image shows multiple hamartomas and a large subependymal nodule. |
MRI scans often demonstrate subependymal nodules with varying degrees of contrast enhancement. Calcifications are frequent and are best demonstrated using gradient-echo sequences. Cortical tubers, ranging in size from 1 to 2 cm, are located at the gray–white matter interface. The parietal and frontal lobes are affected most often (Fig. 3). Tubers are isointense on T1 images but hyperintense using T2 sequences. Gyral core tubers may resemble an empty gyrus due to hypointensity of the white matter.3,48,129
The location of certain size tubers appears to correlate with EEG epileptogenic foci and prognosis. Patients with posteriorly located lesions have early onset of seizures as opposed to those with frontal lesions.31 The presence of multiple and large cortical tubers, early onset of multiple seizure types, and multifocal EEG abnormalities correlate with unfavorable prognosis.
Lissencephalies
Lissencephaly refers to brains without normal sulcation (i.e., smooth brains).43,72 The lissencephalies are a group of different disorders with distinct pathologic substrates and multiple causes. The major distinction is between classical lissencephaly and cobblestone lissencephaly, terms that reflect the appearance of the brain and that, in turn, derive from different genes. The various types of lissencephaly are classified according to the gene defect and associated malformations. For example, instead of classifying LIS1 mutations and DCX mutations as subcategories of the isolated lissencephaly sequence, the classification includes Miller-Dieker syndrome, isolated lissencephaly syndrome, and subcortical band heterotopia as subcategories under LIS1 mutations.10 Instead of listing POMT1 mutations and FKRP mutations under Walker-Warburg syndrome, the classification lists muscle-eye-brain disease and Walker-Warburg syndrome under FKRP mutations. As a result of this reclassification, many syndromes are listed more than once. For example, isolated lissencephaly sequence and band heterotopia are listed under both LIS1 and DCX mutations and Walker-Warburg syndrome is listed under POMT, FKRP, and FCMD mutations.
Classical Lissencephaly
Classical lissencephaly or generalized agyria-pachygyria is a severe brain malformation manifested by a smooth cerebral surface, abnormally thick cortex with four abnormal layers,
diffuse neuronal heterotopia, enlarged ventricles, and often hypoplasia of the corpus callosum. Mutations in the LIS1 gene result in Miller-Dieker syndrome (MDS), isolated lissencephaly sequence (ILS), and subcortical band heterotopia (SBH). Similar phenotypes also occur with DCX mutations, although in patients with DCX mutations, the frontal lobes are most affected; whereas in patients with LIS1 mutations, the posterior areas are more involved. Mutations of the ARX gene cause X-linked lissencephaly with ambiguous genitalia and anomalies of the corpus callosum.65,69,70,127
diffuse neuronal heterotopia, enlarged ventricles, and often hypoplasia of the corpus callosum. Mutations in the LIS1 gene result in Miller-Dieker syndrome (MDS), isolated lissencephaly sequence (ILS), and subcortical band heterotopia (SBH). Similar phenotypes also occur with DCX mutations, although in patients with DCX mutations, the frontal lobes are most affected; whereas in patients with LIS1 mutations, the posterior areas are more involved. Mutations of the ARX gene cause X-linked lissencephaly with ambiguous genitalia and anomalies of the corpus callosum.65,69,70,127
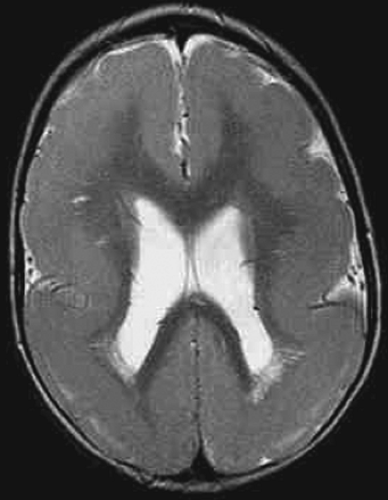 FIGURE 4. Lissencephaly. Miller-Dieker syndrome. LIS1 mutation. Axial T2-weighted image shows smooth cortex. |
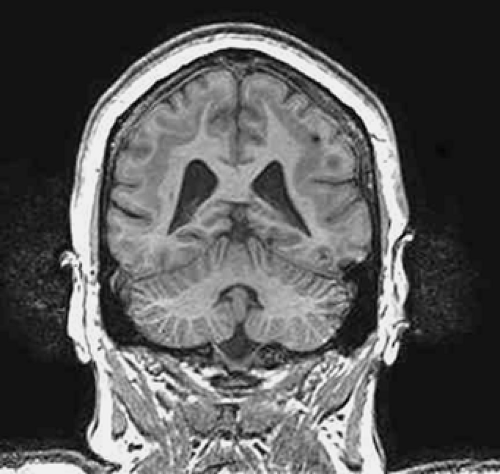 FIGURE 5. Subcortical band heterotopia (SBH). Coronal T1-weighted image shows typical subcortical band of gray matter with relative normal cortical infolding; DCX mutation. |
Children with classical lissencephaly present with feeding difficulties or hypotonia. By 6 months of life, most of these children will have seizures, and the evolution of the epilepsy is similar in all patients. Infantile spasms with hypsarrhythmia and typical paroxysmal fast activity on the EEG appear in the first year of life. Response to treatment with adrenocorticotropic hormone (ACTH) or other anticonvulsants is variable, but most children will continue to have frequent seizures accompanied by severe developmental delay. Typical seizure types also include myoclonic, tonic, and tonic–clonic seizures. Profound mental retardation and spastic quadriplegia are present.
Diagnosis of classic lissencephaly is based on the typical clinical, EEG, and MRI features. MRI demonstrates a thickened cortex, loss of white matter, and vertical sylvian fissures, which result in the typical 8-shaped appearance of the brain (Fig. 4). Cortical thickness is in the range of 11 to 20 mm compared to 3.5 mm in normal controls.11 In some patients there are regions of pachygyric cortex. Barkovich et al.11 have also reported the presence of incomplete inversion of the hippocampi, a marker of arrest of neuronal migration.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


