Table 26.2 Genetic Etiologies of Malformations of Cortical Development


MALFORMATIONS OF CORTICAL DEVELOPMENT
Accurate diagnosis of MCD relies primarily on recognition of the malformation on brain MRI, which, in turn, determines correct prognosis and genetic counseling. In the following sections, the genetic, imaging, and functional aspects of the most common MCD are discussed, with special emphasis on epilepsy associated with these disorders.
MCD DUE TO ABNORMAL PROLIFERATION/APOPTOSIS (ABNORMALITIES OF BRAIN SIZE)
Malformations in this group are characterized by an increase or decrease in the number of neurons and glia with corresponding changes in brain size, designated as either microcephaly (MIC) or megalencephaly (MEG). The most common types of MIC and MEG are not typically included under brain malformations because brain structure appears grossly normal in isolated forms. However, detailed studies of neuronal cell types are not available. Furthermore, MIC and MEG often coexist with other developmental brain disorders such as polymicrogyria (PMG) and periventricular nodular heterotopia (PNH) in distinct phenotypes.
Microcephaly Syndromes
MIC is defined as head circumference 3 standard deviations (SDs) or more below the mean for the individual’s age and gender. Historically, a confusing combination of terms has been used to describe and classify different types of MIC. When severe congenital MIC is the only abnormality on evaluation, that is, without other brain or somatic abnormalities, the terms primary MIC and microcephaly vera have been historically used though these terms are often poorly understood (4). Most patients with congenital MIC fall broadly into two clinical groups (5). The first group comprises children with extreme MIC but only moderate neurologic problems, usually moderate intellectual disability (ID) without spasticity or epilepsy. Several genes associated with this phenotype have been identified (see Table 26.2). The second and more important group from an epilepsy standpoint consists of congenital MIC with severe spasticity and epilepsy (6). Children in this group present with severe ID, spastic quadriparesis, gastroesophageal reflux, and poor feeding, leading to failure to thrive. Early-onset intractable epilepsy is common. Brain MRI universally shows a simplified gyral pattern. Other neuroimaging features in this group include increased extra-axial space, delayed myelination, agenesis of the corpus callosum, and disproportionate hypoplasia of the brainstem and/or cerebellum. This clinical spectrum constitutes pathogenetically heterogeneous conditions, as several genes have now been identified in this group (see Table 26.2).
Children with severe congenital MIC are often erroneously diagnosed with lissencephaly (LIS) because of reduced number of broad gyri. However, the cortex is not as thick as in true LIS (Fig. 26.1). The term microlissencephaly applies to the few patients with severe congenital MIC and a truly thickened cortex, and these patients also usually present with intractable epilepsy. By far, the majority of congenital MIC syndromes are inherited in an autosomal recessive manner.
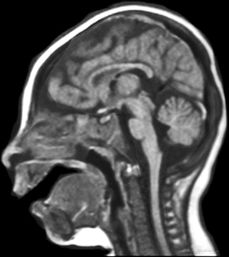
Figure 26.1. Microcephaly. Sagittal MRI demonstrates MIC (<3 SD) with relative preservation of normal sulcal cerebral anatomy.
Postnatal MIC, on the other hand, is characterized by borderline small head size (2 to 3 SD below the mean) at birth that later progresses. Postnatal MIC is a feature of more than 300 developmental syndromes that are beyond the scope of this chapter.
Megalencephaly Syndromes
MEG is defined as an oversized and overweight brain that exceeds the mean by 2 or 3 SD for age and gender. MEG may occur as a mild familial variant with normal brain structure or as part of a growing number of genetic syndromes, including metabolic and nonmetabolic disorders (6). The clinical findings of individuals with MEG vary, but neurologic problems are usually mild to moderate, particularly in the familial form. A subset of patients have severe ID, intractable epilepsy, and other neurologic abnormalities. These severe outcomes are related to the presence of other cortical brain abnormalities, such as PMG. The most common MEG syndromes are associated with characteristic somatic and neuroimaging features (see Table 26.2).
MCD DUE TO ABNORMAL PROLIFERATION (ABNORMAL CELL TYPES)
Malformations in this group are characterized by abnormal neurons and, often, glia as well. These are usually localized malformations. In some patients, abnormal cell types have been classified as neoplastic, although the malignant potential is low. The most common of these is tuberous sclerosis complex (TSC) (reviewed in Chapter 30).
Hemimegalencephaly
Hemimegalencephaly (HMEG) is a brain malformation characterized by an enlarged and dysplastic cerebral hemisphere (Fig. 26.2). The overgrowth and dysplasia may involve an entire hemisphere, part of a hemisphere, and/or part of the contralateral hemisphere as well. Macroscopically, the involved hemisphere is enlarged with cortical dysgenesis, white matter hypertrophy, and a dilated and dysmorphic lateral ventricle. There is no clear predilection for right or left sides. The microscopic features of HMEG vary significantly and may include PMG, heterotopic gray matter, cortical dysplasia (cortical dyslamination, bizarre enlarged neurons, balloon cells), blurring of the gray–white junction, and increase in the number of neurons and astrocytes (7).
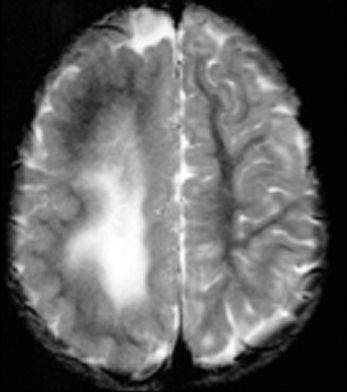
Figure 26.2. Hemimegalencephaly. Axial MRI shows large hemisphere with white matter changes. Note the smooth cortex in the posterior region.
HMEG is most often isolated but can be sporadically associated with neurocutaneous syndromes such as linear nevus sebaceous syndrome, hypomelanosis of Ito, TSC, and neurofibromatosis. HMEG with somatic overgrowth has been reported in Klippel–Trenaunay syndrome and Proteus syndrome (6). Postzygotic activating mutations in components of the phosphatidylinositol-3-kinase (PI3K)-v-akt murine thymoma viral oncogene homolog (AKT) pathway have been identified in individuals with HMEG (8). Mutations of the same genes have been identified in children with MEG syndromes, such as the megalencephaly capillary malformation (MCAP) and the megalencephaly–polymicrogyria–postaxial polydactyly–hydrocephalus (MPPH) syndromes (9). These mutations lead to gain of function and activation of the PI3K-AKT pathway, a critical cellular pathway that regulates diverse cellular functions such as cell growth, proliferation, survival, apoptosis, tumorigenesis, and brain development.
The classic clinical triad of HMEG is (i) intractable partial seizures with onset in the neonatal period or early infancy, (ii) unilateral or focal neurologic signs (hemiparesis, hemianopia), and (iii) intellectual disability. Seizures are typically partial and almost always intractable to medical therapy. Infantile spasms, tonic seizures, or electroclinical features of Ohtahara syndrome or West syndrome may occur (10).
The MRI appearance of HMEG is characteristic. Most affected individuals have moderate to severe enlargement of one cerebral hemisphere. In some, enlargement may be localized to the frontal or temporoparietal regions, but in others, it may extend to distinct regions of the contralateral hemisphere. Gray matter is almost uniformly abnormal showing areas of thickening and simplification or overfolding, resembling pachygyria or PMG, respectively. The underlying hemispheric white matter may be increased or decreased, with abnormal signal characteristics in some patients. Heterotopia is commonly seen, and the ventricular system is enlarged and/or dysplastic in most patients. Electroencephalographic abnormalities are often broadly distributed throughout the abnormal hemisphere, and most severe cases exhibit a suppression burst pattern early on.
Predictors of poor outcome in HMEG are severity of hemiparesis, smoothness of the cortical surface on MRI, and abnormal activity on electroencephalography (EEG). Anatomical or functional hemispherectomy may improve both epilepsy and ID in selected patients (11). However, some patients do poorly with hemispheric surgery, possibly due to more widespread but asymmetric malformations.
Focal Cortical Dysplasia
The term focal cortical dysplasia (FCD) designates a spectrum of abnormalities of the laminar structure of the cortex, variably associated with cytopathologic features including giant (or cytomegalic) neurons, dysmorphic neurons, and balloon cells (Fig. 26.3). There have been various attempts to classify FCD based on subtle histologic characteristics, and the most recent classification considers clinical presentation and imaging findings, in addition to histopathologic features (12). According to this system, FCD type I refers to isolated lesions, which present either as radial (type Ia) or as tangential (type Ib) dyslamination of the neocortex, microscopically identified in one or multiple lobes. FCD type II is an isolated lesion characterized by cortical dyslamination and dysmorphic neurons without (type IIa) or with balloon cells (type IIb). FCD type III occurs in combination with hippocampal sclerosis (type IIIa) or with epilepsy-associated tumors (type IIIb). FCD type IIIc is found adjacent to vascular malformations, whereas FCD type IIId can be diagnosed in association with epileptogenic lesions acquired in early life (i.e., traumatic injury, ischemic injury, or encephalitis). The pathologic features of the tubers of TSC and FCD type IIb have significant overlap.
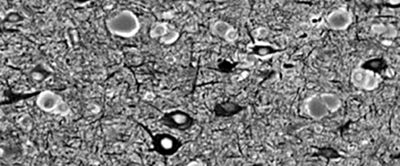
Figure 26.3. Focal cortical dysplasia. Silver staining showing irregular arrangement of big neurons and pale brown balloon cells.
According to the prevailing hypothesis, FCD originates from abnormal migration, maturation, and cell death during ontogenesis (13). The close cytoarchitectural similarities between FCD and cortical tubers of TSC prompted the hypothesis of a common pathogenetic basis, and a study has supported the role of the TSC1 gene in the pathogenesis of FCD, although these data remain to be further substantiated (14). In one paper, human papillomavirus (HPV) infection was implicated in FCD type IIB. The specific HPV 16 oncoprotein E6 (HPV16 E6) is a potent activator of mTORC1 signaling, further suggesting a relationship between FCD and the PI3K-AKT-mTOR pathway (15). Importantly, histopathologic similarities between FCD, HMEG, and the dysembryoplastic neuroepithelial tumors, two highly epileptogenic developmental lesions, further support the hypothesis of a developmental origin. Finally, FCD has been postulated to be linked to perinatal or early postnatal brain injury, with subsequent cell differentiation in the scarred area.
The most common clinical sequelae of FCD are seizures, ID, and focal neurologic deficits. Epilepsy is usually focal, intractable, and often complicated by focal status epilepticus. FCD has been shown to be intrinsically epileptogenic in vivo using electrocorticography during epilepsy surgery and in vitro using cortex resected from patients with intractable epilepsy (16,17).
FCD is rarely visible by computerized tomography (CT), and the mildest malformations may not be visible on current MRIs. Other lesions can be detected by blurring of the cortex–white matter junction on T1-weighted images as well as cortical thickening or abnormal T2 or fluid-attenuated inversion recovery hyperintensity in the white matter of a gyrus or in the depth of a sulcus. The term transmantle dysplasia applies to a band of abnormal signal intensity that extends from the cortex to the superolateral margin of the lateral ventricle (Fig. 26.4). Another subtype of FCD is bottom of the sulcus focal cortical dysplasia (18). This type of FCD can be very small in size and may be easily overlooked on imaging. However, once diagnosed, patients may benefit greatly from surgery as the lesions can often be resected in total.
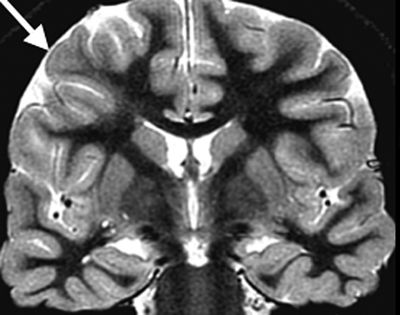
Figure 26.4. Coronal T2W MRI showing an area of irregular cortical folding (arrow) with blurring of the gray–white matter junction and underlying increased signal intensity in the white matter, extending from the subcortex to the ventricular wall. This combination of findings is consistent with FCD.
Cortical Dysplasia with Neoplastic Changes
Several low-grade, primarily neuronal, neoplasms are associated with cortical dysplasia, including dysembryoplastic neuroepithelial tumors, ganglioglioma, and gangliocytoma. Controversy continues over their proper classification. These neoplasms occur most often in children and young adults, and the frequency of these neoplasms in epilepsy surgical series is approximately 5% to 8% (19). Tumors are often located in the temporal lobes, where residual heterotopic neurons in the white matter are also common but can also present in other brain regions. While patients usually exhibit partial seizures that are difficult to control with medication, complete resection of the lesion may lead to seizure freedom.
MALFORMATIONS DUE TO ABNORMAL NEURONAL MIGRATION (NEURONAL MIGRATION DISORDERS)
Lissencephaly and Subcortical Band Heterotopia
LIS is characterized by absent (agyria) or decreased (pachygyria) convolutions, producing cortical thickness and a smooth cerebral surface. Several types of LIS have been recognized. The most common, classical (or type 1) LIS, features a very thick cortex (10 to 20 mm as compared to a normal thickness of 4 mm) without other brain malformations. The cytoarchitecture consists of four primitive cortical layers, rather than the normal six. From the cortical surface inward, these consist of a (i) poorly defined marginal zone with increased cellularity; (ii) superficial cortical gray zone with diffusely scattered neurons; (iii) relatively neuron-sparse zone; and (iv) deep cortical gray zone with neurons often oriented in columns (20, 21).
Subcortical band heterotopia (SBH) consists of bands of gray matter interposed in the white matter between the cortex and the lateral ventricles (Fig. 26.5) (6). LIS and SBH are MCD that manifest along the same spectrum. This conclusion is based on observations of rare patients with areas of LIS that merge into SBH and of multiple families with X-linked LIS in males and SBH in females.
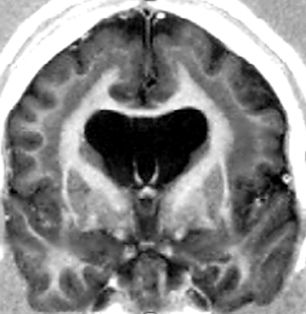
Figure 26.5. SBH. DCX mutation in a female. Coronal T1W image shows typical SBH with relative preservation of cortical anatomy.
The two most common genes associated with classic LIS and SBH are LIS1 and DCX. LIS1 (PAFAH1B1) is responsible for the autosomal form of LIS1, while the doublecortin (DCX) gene is X-linked. Although either gene can result in LIS or SBH, most cases of classic LIS are due to deletions or mutations of LIS1, whereas most cases of SBH are due to mutations of DCX. LIS1-related LIS is more severe in the posterior (p) brain regions (p > a gradient), whereas DCX-related LIS is more severe in the anterior (a) brain (a > p gradient) (Fig. 26.6).
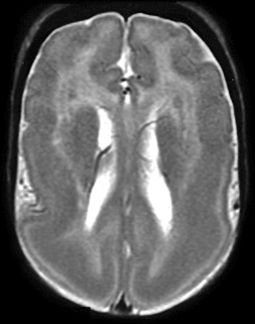
Figure 26.6. Lissencephaly. LIS1 mutation. MRI shows the typical smooth cortex predominantly affecting the posterior regions.
Children with classic LIS often appear normal as newborns but may present with apnea, poor feeding, or hypotonia. Seizures are uncommon during the first few days of life but typically begin before 6 months of age, and the clinical presentation is often similar. Infantile spasms (ISS) occur in 80% of affected children within the first year of life, often appearing initially as hypsarrhythmia on EEG. ISS respond to ACTH or other anticonvulsants in the majority of patients, but in the long term, almost all affected children have frequent seizures and many meet criteria for Lennox–Gastaut syndrome. Profound ID, hypotonia, mild spastic quadriplegia, and opisthotonus are also seen. Many patients require a gastrostomy because of poor nutrition and repeated episodes of aspiration and pneumonia.
In contrast, patients with SBH and the rare patients with partial LIS have mild to moderate ID (although normal intelligence or severe ID has been reported), minimal pyramidal signs, and dysarthria (22). Seizures usually begin during childhood but may appear later in life. Multiple seizures types may occur that can be difficult to control; however, the frequency and severity vary among affected individuals. As with other developmental syndromes, epilepsy may be an independent factor to cognitive delay and overall neurodevelopmental status. EEGs usually show generalized spike–wave discharges or multifocal abnormalities. The neurologic outcome depends on the thickness of the heterotopic band on MRI and associated malformations.
Lissencephaly Syndromes and Genes
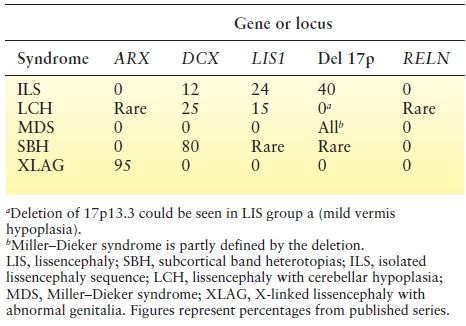
The most common LIS syndromes include (i) isolated LIS sequence (caused by DCX in males; LIS1; and, rarely, TUBA1A, TUBB2B, TUBG1, and DYNC1H1), (ii) SBH (DCX in females and rare in males, and LIS1), (iii) Miller–Dieker syndrome (MDS) (contiguous deletion of LIS1 and YWHAE), (iv) several types of LIS with cerebellar hypoplasia (LCH) (RELN and VLDLR), (v) X-linked LIS with abnormal genitalia (XLAG) (ARX), and (vi) Baraitser–Winter syndrome (BWS) (ACTB1 and ACTG1), among others. Table 26.3 lists the frequency of mutations in these syndromes. Careful review of brain imaging and clinical features can distinguish these syndromes and helps with the identification of the causative gene.
Table 26.3 Frequency of Mutations in LIS and SBH Syndromes
Isolated lissencephaly sequence (ILS) consists of classic LIS with mild facial dysmorphism including mild bitemporal hallowing and small jaw (23). ILS associated with mutations of the X-linked DCX gene is characterized by either a severe LIS with no apparent gradient or an a > p gradient and normal facial appearance, whereas mutations or deletions of the LIS1 gene produce LIS with p > a gradient (see Fig. 26.6). The facial appearance may be normal or involve subtle dysmorphism similar to but milder than MDS. Mutations of the tubulin genes (TUBA1A, TUBB2B, TUBG1) are characterized by LIS in addition to other complex developmental brain abnormalities such as PMG, callosal hypoplasia or agenesis, abnormal basal ganglia, and cerebellar hypoplasia (see Table 26.2) (24-27).
MDS is the prototypic LIS syndrome associated with characteristic facial features and other birth defects such as heart malformations and omphalocele. Facial features include prominent forehead, bitemporal narrowing, short nose with upturned nares, protuberant upper lip with a thin vermilion border, and small jaw. LIS in MDS is severe with no apparent gradient or rarely a p > a gradient similar to ILS with LIS mutations. All patients have deletions of chromosome 17p13 that include LIS1 and YWHAE. About 60% to 70% of deletions are detected by karyotype, and the remainder are detectable by fluorescence in situ hybridization or chromosomal microarray (28-31).
LCH affects a small percentage of patients with LIS syndromes. Group a, the most common type, resembles isolated LIS syndrome but with mild cerebellar vermis hypoplasia. Some patients have mutations of DCX or LIS1 but much less frequently than patients with typical ILS. Group b consists of moderate LIS with an a > p gradient, moderate 8- to 10-mm cortical thickness, a globular hippocampus, and a small afoliar cerebellum. Mutations in RELN and VLDLR have been identified in individuals with LCH overall.
XLAG is a variant LIS characterized by a p > a gradient, intermediate 8 to 10 mm cortical thickness, total agenesis of the corpus callosum, often cavitated or indistinct basal ganglia, severe postnatal MIC, and ambiguous or severely hypoplastic genitalia. Affected children have profound ID, hypothalamic dysfunction with poor temperature regulation, intractable epilepsy typically beginning on the first day of life, infancy-onset dyskinesia that may be difficult to distinguish from seizures, and chronic diarrhea. Female relatives, including mothers, have isolated agenesis of the corpus callosum. Mutations of the ARX gene have been found in almost all patients with XLAG.
BWS is a rare but recognizable disorder characterized by congenital ptosis, high-arched eyebrows, hypertelorism, ocular colobomata, and anterior predominant LIS. Other typical features include postnatal short stature, MIC, ID, seizures, and hearing loss. Recently, de novo mutations in ACTB and ACTG1 have been identified in individuals with BWS (see Table 26.2) (32).
Cobblestone Brain Malformations (Cobblestone Complex)
The cobblestone complex (previously known as type 2 or cobblestone LIS) is a severe brain malformation consisting of cobblestone cortex, abnormal white matter, enlarged ventricles often with hydrocephalus, small brainstem, and small dysplastic cerebellum (Fig. 26.7). In the most severely affected patients, the brain surface is smooth, which previously led to the designation LIS, although less severe cobblestone malformations have an irregular, pebbled surface rather than a smooth surface. Severely affected individuals may have progressive hydrocephalus, large posterior fossa cysts (atypical for Dandy–Walker malformation), and occipital cephaloceles. Eye malformations are frequent, and congenital muscular dystrophy is probably always present.
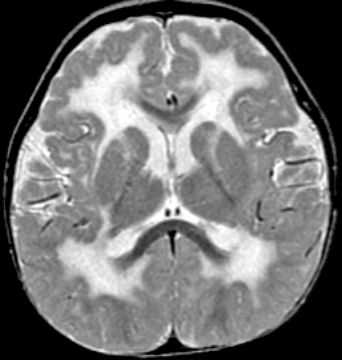
Figure 26.7. Axial T2W MRI shows extensive white matter changes and PMG typical of cobblestone malformation due to a Fukutin mutation.
Related posts:
 Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Dietary Therapies for Epilepsy
Epidemiologic Aspects of Epilepsy
Application of Electroencephalography in the Intensive Care Setting
Hormones, Catamenial Epilepsy, Sexual Function, and Reproductive and Bone Health in Epilepsy
Other Nonepileptic Paroxysmal Disorders
Driving and Social Issues in Epilepsy
Dietary Therapies for Epilepsy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


