Mechanisms of Drug Resistance
Sanjay M. Sisodiya
Heinz Beck
Wolfgang Löscher
Annamaria Vezzani
Introduction
For most patients with a diagnosis of epilepsy requiring drug treatment, the outlook is favorable. In mature economies, about 70% of patients can expect to become seizure free with antiepileptic drug (AED) treatment.69 This percentage is an overall figure and may differ for subgroups, and most probably depends on etiology and syndrome.33,72 Although some patients undoubtedly have complex response patterns that change over time for no obvious reason and defy categorization, some 30% of patients will never have any sustained period of seizure freedom—for example, >12 months—in the history of their epilepsy. Scientific inquiry into the concepts and mechanisms of such resistance to AED treatment has gained significant momentum recently. The resulting body of work constitutes the subject of this chapter.
Epilepsy is a heterogeneous group of conditions. The pathophysiologic derangements in the brain are complex and not fully understood for any single seizure type or epilepsy syndrome. There are only a limited number of ways in which the brain, despite its complexity, can actually manifest its function or dysfunction: Epilepsy and seizures are final common manifestations of a wide range of disruptions in brain structure and function. Of course, many of these disruptions share commonalities at various levels, and advantage is taken of these shared characteristics in labeling, understanding, diagnosing, and treating “epilepsy.” Both shared and syndrome-specific characteristics might complicate and hamper these same efforts at managing seizures and epilepsy in humans. As a biologic and clinical phenomenon, resistance to AED treatment is one such shared, complex, multifactorial, and adaptive aspect of epilepsy biology. The study of drug resistance might therefore not only improve rational treatment for patients with drug-resistant epilepsy, with its associated morbidity and mortality, but also cast light on disease pathophysiology, determinants of natural history, and predictors of prognosis.
Recent comprehensive reviews have thoughtfully discussed not only the biologic bases of refractory epilepsy, but also the definition of such epilepsy.32,72 There may be two patients with the same demography, the same syndrome, and the same underlying imaging and histologic pathology, one of whom responds to the first AED tried and the other who never responds with any prolonged period of seizure freedom despite years of administration of a series of differing AEDs. The effect of AEDs on prognosis in an individual is unknown.32 AEDs generally have a limited spectrum of potential mechanisms of action, and they may undoubtedly render some individuals seizure free, a state associated with proven benefits. Therefore the question of why some individuals have epilepsy refractory to currently available AEDs is both valid and important.
As an illustration of some of these concepts, it is worthwhile considering epilepsy caused by a particular neurodevelopmental abnormality, periventricular heterotopia (PVH) caused by mutation in the X-linked gene FLNA, encoding filamin A. Deleterious mutations in this gene can underlie some cases of familial or sporadic PVH:21 In some cases, the PVH goes on to cause epilepsy, which may be drug resistant, whereas other patients do not even develop epilepsy.22 We have a reasonable understanding of the role of filamin in neurodevelopment.76 We know much about the pathologic anatomy of periventricular nodules themselves, including the disruptions of neurohistologic and immunophenotypic profiles, the abnormal electrical activity that may arise from such nodules, their connectivity with the rest of the brain, and their appearance on imaging. It is clear that even following a single pathologic nucleotide alteration in one gene causing epilepsy as its primary manifestation, a tangled multilayered raft of changes occurs at multiple temporal, spatial, and functional time points, leading eventually in a proportion of cases to drug-resistant epilepsy. Most cases of refractory epilepsy are due to more common pathologies, such as hippocampal sclerosis, but remain in some ways less well understood at root, even though they have been longer studied. It is therefore unsurprising that the biologic bases of AED resistance should be so poorly comprehended.
There are two main hypotheses for the underlying cause of refractory, or drug-resistant, epilepsy: (a) the “target” and (b) the “transport” hypotheses. In essence, the former focuses on pharmacodynamic resistance involving the postulated mechanism of AED action; the latter hinges on pharmacokinetic resistance, positing that overactive transport mechanisms prevent AEDs from reaching their targets in sufficient concentrations in the first place. These hypotheses will be considered in greater detail. Eventually, a number of mechanisms are likely to emerge as important to greater or lesser extents in a range of syndromes. Some other potential mechanisms are also considered.
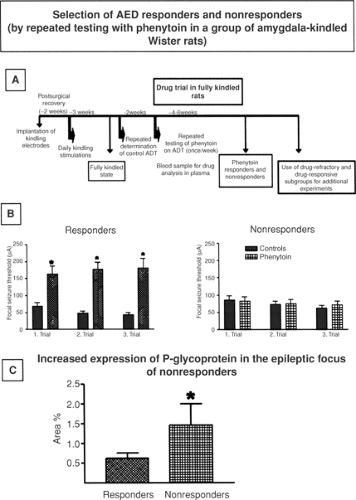 FIGURE 1. Selection of responders and nonresponders to antiepileptic drug (AED) treatment in the kindling model of temporal lobe epilepsy. A: The experimental protocol used to select responders and nonresponders from a large group (usually >50) of amygdala-kindled Wistar rats. ADT, afterdischarge threshold. B: The results of a typical selection experiment. Responders always react with a significant increase of focal seizure threshold in each trial with administration of phenytoin, whereas nonresponders do not show any anticonvulsant effect of phenytoin at the same doses. Results shown are means and SEM, *p <.05. C: The expression of the drug efflux transporter P-glycoprotein (P-gp) in brain capillary endothelial cells that form the blood–brain barrier. Nonresponders exhibit significantly higher expression of P-gp than do responders in the epileptic focus, that is, the kindled amygdala. The expression is shown as area of brain capillary endothelial cells immunohistochemically stained for P-gp. This area was determined by computer-assisted image analysis of brain sections. Data are from Brandt C, Bethmann K, Gastens AM, et al. The multidrug transporter hypothesis of drug resistance in epilepsy: Proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006;24(1):202–211. |
Animal Models of Drug-Resistant Epilepsy
Animal models of epilepsy allowing selection of pharmacoresistant and pharmacosensitive subgroups of animals constitute a valuable tool for studying mechanisms of intractability and developing more effective treatment strategies.36,38 Only one model with these characteristics has been extensively explored: The amygdala-kindled Wistar rat, from which phenytoin-resistant and phenytoin-sensitive rats can be selected by repeated testing with phenytoin (Fig. 1).36,38 Phenytoin-resistant rats are also resistant to various other AEDs, thus offering parallels with the clinical presentation of multidrug-resistant temporal lobe epilepsy. Phenytoin-resistant kindled rats have been used to study mechanisms of drug resistance, described in more detail later. In short, breeding and other studies have indicated that genetic factors and kindling-associated brain alterations are involved in drug resistance in this model.37 However, one major drawback of this model is that kindled
rats do not exhibit spontaneous recurrent seizures, so that elicited seizures have to be used for drug studies. This is also true for the 6-Hz “psychomotor” seizure model in mice, which has been proposed as a useful model of therapy-resistant limbic seizures.3
rats do not exhibit spontaneous recurrent seizures, so that elicited seizures have to be used for drug studies. This is also true for the 6-Hz “psychomotor” seizure model in mice, which has been proposed as a useful model of therapy-resistant limbic seizures.3
Some recent studies have indicated that rats with spontaneous recurrent seizures developing after status epilepticus differ in their individual response to AEDs, thus allowing selection of drug-resistant and drug-responsive subgroups.8,19 This group of models reflects most closely a common form of human drug-resistant epilepsy, the mesial temporal lobe epilepsy (mTLE) syndrome. By using drug-resistant epileptic rats selected from such a post–status epilepticus model of mTLE, it was recently shown that drug-resistant rats differ from drug-responsive rats in (a) expression of multidrug efflux transporters in the brain,99 (b) expression of drug targets in the brain,97 and (c) morphologic alterations in the hippocampus.97 Thus, such post–status epilepticus models, allowing selection of drug responders and nonresponders, are interesting tools for studying mechanisms of drug resistance. As reported recently, they can also be used to study whether overcoming the mechanism(s) counteracts drug resistance.6
Apart from selecting subgroups of drug-resistant rats from models of mTLE, another approach is to use models in which all animals appear per se resistant to treatment with certain AEDs.38 Such models include the 6-Hz “psychomotor” seizure model in mice,3 the MAM (methylazoxymethanol acetate) model of cortical dysplasia in rats,82 and certain post–status epilepticus models of TLE.20 Furthermore, several in vitro brain slice preparations have been proposed to be useful in studying mechanisms of drug resistance.38,105 As with all models of drug-resistant epilepsy, one major problem in validating these in vitro models is the lack of a positive control, that is, a novel AED with clinical efficacy in the management of refractory epilepsy. As a consequence, it is not known whether novel compounds that are found to be active in models of drug-resistant seizures will be subsequently found to be effective for the management of human refractory epilepsy.104
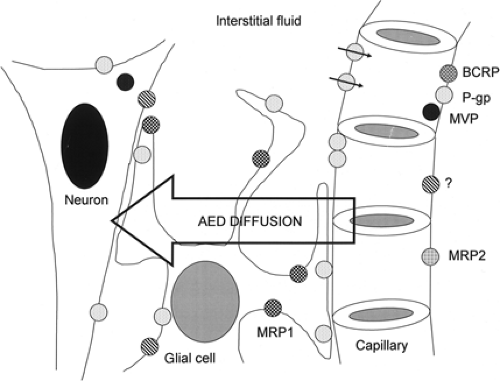 FIGURE 2. Schematic illustrating the transport hypothesis. Most antiepileptic drugs (AEDs) diffuse from the capillary lumen to the interstitial fluid and their targets, usually on the surface of or within neurons. Nonspecific, multidrug transporters (represented by variously filled circles) are present in particular distributions for given transporters. Thus, for example, multidrug-resistance–associated protein 1 (MRP1, checkered circles) is overexpressed on glial cells in epileptogenic tissue. The transport hypothesis maintains that AED concentration at the neuron is reduced by the transport activity of various transporters on capillary endothelial cells, glia, and neurons, reducing diffusion of AEDs to their desired site of action and thus reducing their antiepileptic effects. The circle marked with a question mark has been added to emphasize that we do not yet know all the transporters that might be relevant. BCRP, breast cancer resistance protein; MVP, major vault protein; P-gp, P-glycoprotein. |
Drug Transport Hypothesis
Background
The “transport” hypothesis embodies the concept that sustained failure of AEDs to stop seizures is due to a subtherapeutic local concentration of AEDs at their site of action caused by the activity of multidrug transporters (Fig. 2). The idea arose by analogy with drug resistance in oncology:32,47,89 The in vitro dissection of the mechanisms of resistance of cultured tumor
cells to anticancer drugs showed that the activity of the archetypal transport protein, P-glycoprotein (P-gp, where “P” stands for permeability), was important in cell survival. Extensive subsequent studies in cancerous cells both in vivo and in vitro have revealed a spectrum of transport or transport-related molecules with more or less proven roles in the flux or sequestration of anticancer drugs. Tishler et al. in 199589 were the first to explore this possible mechanism in drug-resistant human epilepsy, showing both increased expression of P-gp in resected epileptogenic human temporal lobe tissue and reduced accumulation of labeled phenytoin in cultured cells artificially expressing P-gp in comparison to the parent wild-type cells not expressing P-gp.
cells to anticancer drugs showed that the activity of the archetypal transport protein, P-glycoprotein (P-gp, where “P” stands for permeability), was important in cell survival. Extensive subsequent studies in cancerous cells both in vivo and in vitro have revealed a spectrum of transport or transport-related molecules with more or less proven roles in the flux or sequestration of anticancer drugs. Tishler et al. in 199589 were the first to explore this possible mechanism in drug-resistant human epilepsy, showing both increased expression of P-gp in resected epileptogenic human temporal lobe tissue and reduced accumulation of labeled phenytoin in cultured cells artificially expressing P-gp in comparison to the parent wild-type cells not expressing P-gp.
The field has advanced considerably since. In oncology, the field has somewhat stagnated after initial excitement that modulation of the activity of transporters might offer dramatic new treatment options in refractory cancer; human studies failed to bear out this optimism. Belatedly, oncologists have established that a number of reasons render questionable the negative results obtained in early studies. For example, the presence of transporters mediating resistance was often presumed and not proven in the particular tumors in particular patients; resistance itself was presumed to be present when it may not have been in all cases involved in trials of inhibitors; the inhibitors used were usually preexisting compounds pressed into service but that had poor inhibitor potency or marked interactions with anticancer agents; and when inhibitors were used, absence of surrogate markers of inhibition meant that the degree to which successful inhibition of transporters in tumoral tissue had been achieved in individual patients was not known.4 Despite these realizations, the area of study has become somewhat discredited (indeed, it is interesting to note that oncology has largely moved to developing specific therapies for specific targets, e.g., herceptin, trasmuzutab24). In epilepsy research, although parallels with cancer should not be drawn too far, we would do well to learn from the experience in oncology.47
Critically, it is important that work on the transporter hypothesis is pursued rationally. One approach is to consider criteria that need to be satisfied to accept a particular transport mechanism as contributing to resistance in human epilepsy. One set postulated is as follows:80
Mechanisms must be detectable in epileptogenic brain tissue
Mechanisms must have appropriate functionality
Mechanisms must actually be active in drug resistance
Overcoming the mechanisms should affect drug resistance
Considerable experimental data exist addressing criteria 1 to 3, reviewed below. Criterion 4 will remain the most demanding, complicated particularly by natural redundancy among the many known and unknown transporters, whose presumptive evolutionary purpose includes the protection of cells, organs, and organisms from environmental or endogenous toxins.
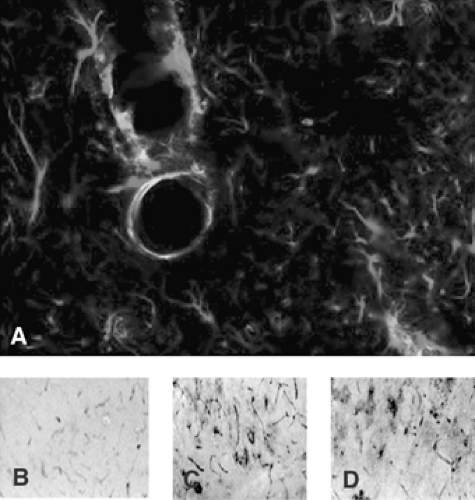 FIGURE 3. Overexpression of P-glycoprotein (P-gp) in an animal model. A: Merged image (20×) showing P-gp immunolabeling (green) and glial fibrillar acidic protein (GFAP)-positive astrocytes (red) in the rat cortex 18 hours after pilocarpine-induced status epilepticus. Colocalization signal (yellow) depicts P-gp expression in astrocytic endfeet adjacent to brain microvessels. B, C, D: Representative immunohistochemical micrographs (10×) depicting P-gp immunolabeling in control rat hippocampus (B) and 18 hours after pilocarpine-induced status epilepticus. Note that in the control panel only, vessels are lightly labeled, whereas seizures induced P-gp overexpression both in vessels and in astrocytes (C, D). (See the color insert.) |
Animal Data: Multidrug Transporter Proteins in Experimental Models of Seizures
Expression Studies
Experimental models of seizures mimicking the neuropathologic features of drug-resistant TLE have been used for studying the molecular mechanisms of intractability.1,11,14,36,73,83 Suitable models should fulfill at least two criteria: (a) the type of seizures should be similar to those occurring in human epilepsy on both behavioral and electrographic manifestations; (b) standard AEDs should be inactive or weakly active as compared to efficacy in models of primarily generalized seizures that are easily suppressed by AEDs. Although the first aspect has been verified in the case of complex partial seizures,36 responsiveness of experimentally induced seizures to AEDs has only rarely been addressed (e.g., in models of status epilepticus evolving to spontaneous seizures83 and in kindled rats36).
P-gp is overexpressed in endothelial cells, and ectopically in astrocytes, after induction of sustained limbic seizures in rodents; neuronal expression has been also reported in some instances.35,75,90,98,106 (Fig. 3). Upregulation of the mRNA of the multiple-drug resistance 1 gene (mdr1, which codes for P-gp) was detected in rat brain during both acute seizures67 and spontaneous seizures90 caused by status epilepticus. After audiogenic seizures in genetically epilepsy-prone rats, P-gp and mdr1 mRNA upregulation was specifically detected in brain regions involved in the onset and propagation of epileptic activity.34 Because limbic seizures were induced in these studies in otherwise normal rodent brain, these experimental findings indicate that epileptic activity per se can increase the expression
of P-gp. These findings are compatible with the observations reported in human TLE tissue (see later discussion).
of P-gp. These findings are compatible with the observations reported in human TLE tissue (see later discussion).
More recently, Volk and Löscher99 reported that phenobarbital-resistant epileptic rats exhibit significantly higher endothelial expression of P-gp in limbic brain regions compared to drug-responsive epileptic rats, providing further support for the multidrug transporter hypothesis of drug-resistant epilepsy. Drug-resistant and responsive rats were selected from a post–status epilepticus model of TLE.99 The severity or duration of status epilepticus or frequency of spontaneous recurrent seizures developing after the status epilepticus did not differ significantly between responders and nonresponders, indicating that drug resistance and overexpression of P-gp may be genetically mediated.
Functional Studies
Rizzi et al.66 reported that mdr1 mRNA is overexpressed in mouse hippocampus after the induction of limbic seizures. When phenytoin was systemically administered to these mice, its brain-to-plasma ratio was 30% less than in mice not subjected to seizures, thus indicating reduced drug concentrations in brain. It is still unknown whether the magnitude of changes in brain phenytoin levels is pharmacologically relevant to reduction of the anticonvulsant activity of this drug, and indeed whether the change in mdr1 mRNA is the unique underlying cause or changes in other, unmeasured entities might also contribute. Potschka et al.53,54 reported that the efficacy of phenytoin in inhibiting generalized clonic seizures in rats was enhanced by probenecid, the nonspecific inhibitor of a subfamily of multidrug transporters, the multidrug-resistance associated proteins (MRPs), and in MRP2-deficient rats; increased efficacy was associated with ∼30% increase in brain-to-plasma concentration ratios of phenytoin. In kindled rats, a widely used model of TLE, significant upregulation of P-gp was reported in brain capillary endothelial cells of limbic brain regions.100 In these rats, brain-to-plasma concentration ratios of phenytoin in the hippocampus were about 30% lower than those measured in control animals.57 When kindled rats were divided into phenytoin responders and nonresponders, nonresponders exhibited a significantly higher expression of P-gp in capillary endothelial cells in the epileptogenic focus (i.e., the kindled amygdala) compared to the same region in responders (FIGURE 1).58
These findings support a link between the level of expression of mdr1 mRNA, P-gp protein or MRP2 protein, and the AED-extrusion function of these drug transporters (e.g., increased mdr1 mRNA/P-gp protein is associated with decreased brain phenytoin levels, whereas decrease in MRP2 protein is associated with increased brain phenytoin levels); in addition, changes in phenytoin brain concentrations of ∼30% appear to affect its efficacy in experimental models.
An important aspect to be considered is the subcellular localization of the ATP-binding cassette (ABC) family of drug transporters, of which P-gp is the archetype.70 Thus, only those proteins localized at the apical membrane of endothelial cells of brain capillaries will be able to decrease whole-brain AED uptake by active extrusion into the blood stream. Examples include P-gp, MRP2, MRP4, and breast cancer resistance protein (BCRP). In contrast, drug transport proteins localized at the basolateral membrane of epithelial cells in the choroid plexus (e.g., MRP1) or in microvasculature (e.g., MRP3 and MRP5) may increase brain uptake of their substrates. Other local changes in the expression of transporters (e.g., in glia) might affect local drug disposition in superimposed local microgradients.
The current hypothesis is, therefore, that P-gp, MRP2, and BCRP localized in microvascular endothelium represent the first barrier for extruding AEDs from the brain. A second barrier may be represented by the presence of P-gp and other transporters in astrocytic endfeet in close apposition with microvasculature. It is notable that parenchymal astrocytes and dysplastic neurons in certain human lesional epileptogenic tissues often express P-gp and MRP1 (see later discussion). The functional consequences of this cell-specific expression are unclear. One speculation is that this multidrug-resistant phenotype in astrocytes and neurons may function as a nonspecific cellular protective mechanism against xenobiotics or toxic compounds that may enter the brain, or be produced in situ, in pathologic conditions. Protective roles for both P-gp28 and MRPs63,64 have been proposed.
Table 1 Drug efflux transporters in central nervous system and antiepileptic drug substrate status | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


