INTRODUCTION
Mitochondria are intracellular organelles ubiquitous amongst eukaryotic cells. Their functions are diverse, but their main responsibility is the production of energy for cellular metabolism. They contain their own DNA, which is inherited along maternal lines, and mutations are a recognized cause of human disease. The involvement of mitochondria in movement disorders is extensive and includes diseases due to mutations of mitochondrial DNA (mtDNA), mutations of nuclear-encoded proteins that are transported to mitochondria, and the effects of mitochondrial toxins, both endogenous and exogenous.
MITOCHONDRIAL STRUCTURE
Mitochondria are double-membraned organelles with an intermembranous space and a matrix that lies within the folded inner membrane. The matrix contains up to several thousand mtDNA molecules. MtDNA is a small circular double-stranded molecule 16,493 bases long encoding two ribosomal RNAs, 22 transfer RNAs, and 13 proteins (Fig. 26.1). These proteins are all part of the OXPHOS system (Table 26.1). MtDNA remains dependent on the nucleus for the production of proteins involved in its transcription, translation, replication, and repair. Thus, although mitochondria have some element of independence in having their own DNA, they remain dependent upon the nucleus for the function of mtDNA and also for the production of all mitochondrial proteins other than the 13 produced by mtDNA.
MtDNA-encoded proteins are translated on mitochondrial ribosomes and incorporated directly into complexes I, III, IV, and V on the inner membrane. Nuclear-encoded proteins are translated on cytosolic ribosomes and are imported into the mitochondrion through a complex receptor and transport system. For many proteins, this involves an N-terminal amino targeting sequence that interacts with membrane receptors and signals the carrier protein’s import into the mitochondrion. The signaling sequence is then cleaved and the protein trafficked to the appropriate mitochondrial compartment. Some proteins are incorporated into the outer mitochondrial membrane and others directly into the intermembranous space.
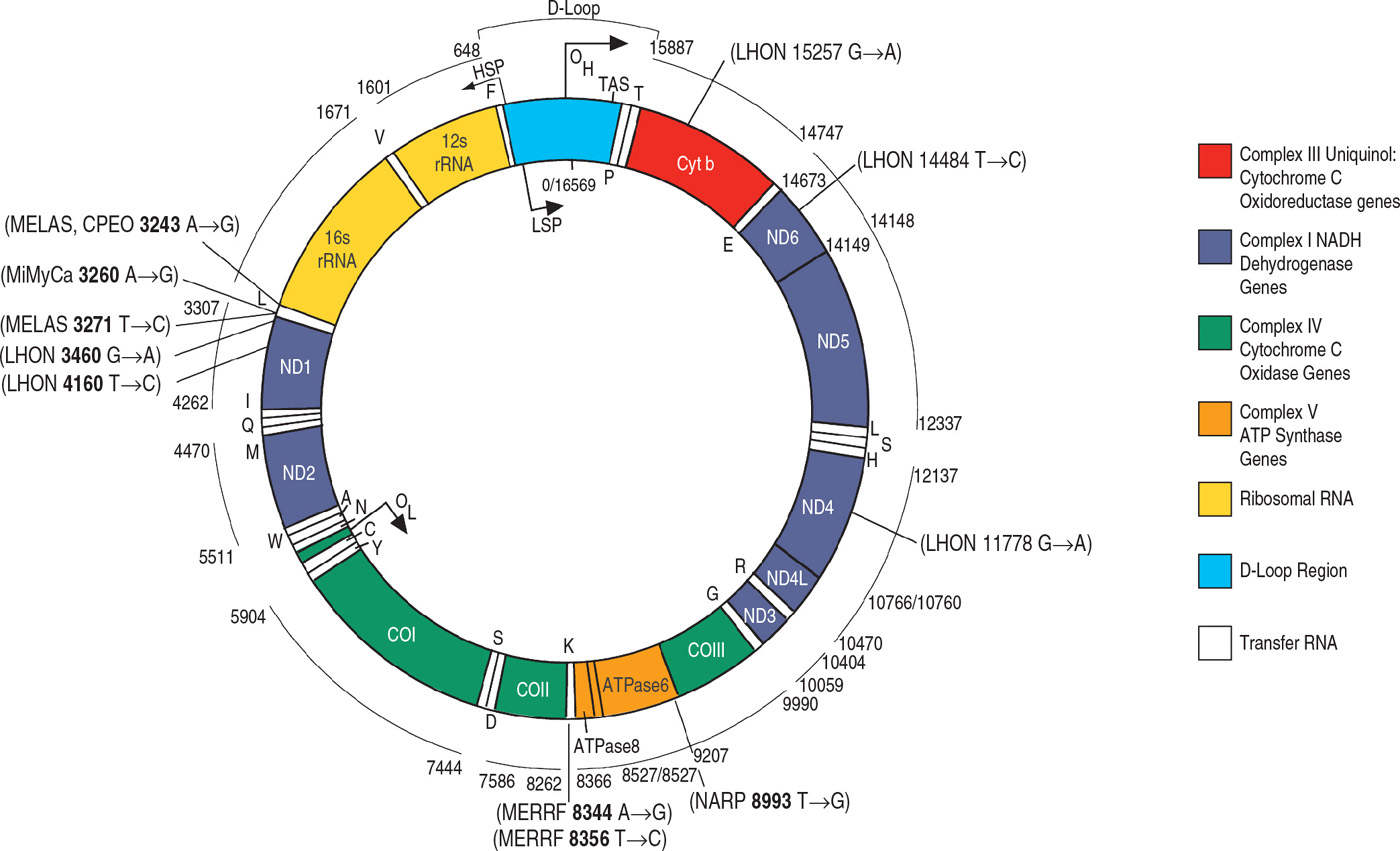
Figure 26.1. Schematic representation of human mitochondrial DNA and the respiratory chain. Included are the genes for respiratory chain proteins and for ribosomal and transfer RNAs. The OH and OL are the origins of heavy- and light-strand replication, respectively. Locations of the more common mitochondrial DNA mutations are indicated.
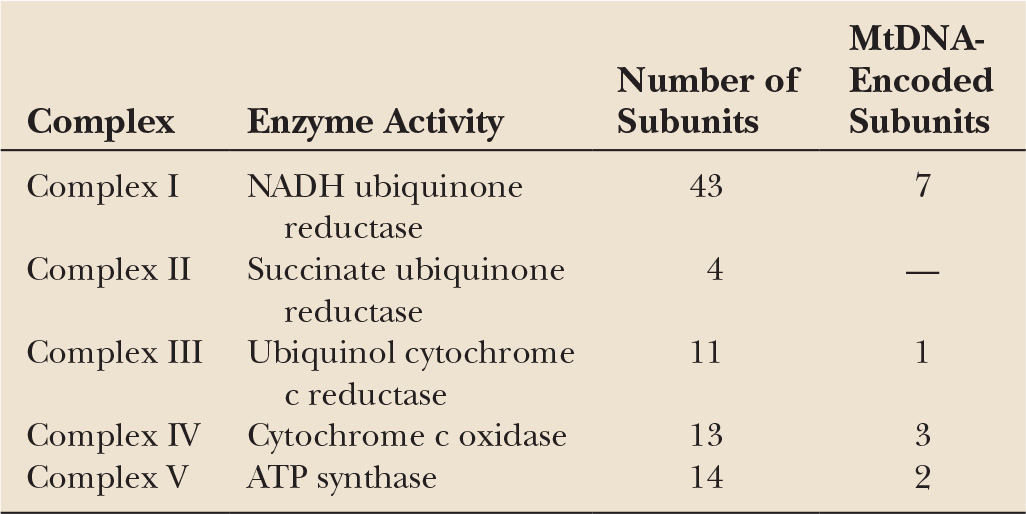
The OXPHOS system is constituted by the four respiratory chain complexes (I–IV) and ATPase (complex V). These proteins are embedded in the inner mitochondrial membrane and comprise a total of approximately 85 subunits, 72 being encoded by nuclear DNA and 13 by mtDNA. The OXPHOS system is not only responsible for ATP production, but is also an important source of superoxide radicals. Defects of the OXPHOS system have the potential not only to cause a failure of energy metabolism but also to increase free radical–mediated damage.
MtDNA continuously replicates, and this function is therefore independent of cell-cycle phases. MtDNA replication involves mtDNA polymerase gamma (POLG), thymidine kinase 2, and deoxyguanosine kinase. MtDNA transcription is necessary for the initiation of replication.
MtDNA is highly polymorphic with numerous differences in sequence between individuals of the same ethnic group, but more so between members of different ethnic groups. MtDNA haplotypes are based upon specific patterns of polymorphisms and appear to have some influence upon the prevalence of certain diseases and the expression of certain mitochondrial mutations, for example, Parkinson’s disease (PD) (see below), senescence, and the expression of Leber’s hereditary optic neuropathy (LHON) mutations.
MtDNA is inherited through the female line, although there has been one report of paternal inheritance of a mtDNA microdeletion in a complex I gene (1). MtDNA mutations are often present in heteroplasmic form, that is coexisting with normal wild-type mtDNA. The proportion of mutant- to wild-type may vary between individuals and between tissues of the same individual. There is a relationship between the mutant load and the degree of biochemical defect, although this is both tissue-dependent and recessive, that is from studies undertaken so far, 5% to 20% of wild-type mtDNA can compensate at the biochemical level.
Mitochondria are randomly segregated at cell division, including oogenesis, and during this process, there is a random segregation or partitioning of wild-type and mutant mtDNA. This results in a varying proportion of wild-type and mutant molecules in daughter cells. When this process occurs through oogenesis, it does so through a “tight bottleneck,” whereby only a small proportion of the mtDNA population is transmitted. A high proportion of mutant load in the oocyte population would result in a high proportion of offspring at risk of developing disease.
The primary function of mitochondria is to support aerobic respiration and to provide energy substrates (ATP) for intracellular metabolic pathways. More recently, mitochondria have been shown to play an important role in cell signaling and particularly in signaling for apoptotic cell death. Mitochondria host a number of metabolic pathways, including the Krebs cycle, β-oxidation, and lipid and cholesterol synthesis. In the context of movement disorders, it is noteworthy that the basal ganglia are sites of high OXPHOS demand, a feature which renders them particularly susceptible to factors that interfere with oxygen delivery directly, for example, hypoxia, or indirectly through the effects of toxins.
MTDNA MUTATIONS AND MOVEMENT DISORDERS
CHRONIC PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA
Chronic progressive external ophthalmoplegia (CPEO) is generally a predominantly myopathic disorder. However, encephalopathic features may also occur with CPEO, and there have been reports of dementia, seizures, myoclonus, and stroke-like episodes (2,3). In a study of 66 patients, 55% had CPEO with muscle weakness but no major CNS disease. Of these, 61% had additional minor features, including salt-and-pepper retinopathy, with preserved acuity in 45%, mild limb or gait ataxia in 41%, extensor plantars and sensorineural deafness were each detected in 23%, and infrequent seizures were found in 5%.
In Kearns–Sayre syndrome (KSS), CPEO is accompanied by pigmentary retinopathy, and one or more of complete heart block, a cerebrospinal fluid (CSF) protein level of above 1 g/L, and ataxia (4). Other cardiac conduction defects described in KSS include left-anterior hemiblock, right bundle branch block, and type II atrioventricular block (5). The diagnostic criteria quote an age of onset below 20 years, but improved identification of cases has revealed later onset in some cases. In pure CPEO, ophthalmoplegia and ptosis may be the only manifestations of this mitochondrial myopathy; however, it is more commonly associated with symptoms such as limb weakness, fatigue, or retinopathy. These cases are often referred to as CPEO plus, and the term KSS reserved for cases in which the above diagnostic criteria are met. In a study of 52 patients, the majority had both ptosis and ophthalmoplegia, with the former occurring in isolation.
MtDNA from CPEO/KSS patients shows large single deletions detectable in DNA extracted from muscle samples, but blood mtDNA analysis usually is normal. These deletions are found in 80% of those with KSS, and 70% of those with CPEO (6–8). MtDNA duplications are also occasionally found in lower abundance (8). Histologic examination of muscle reveals ragged red fibers (RRFs) and a mosaic pattern of cytochrome oxidase (COX)-negative fibers. It is essential that a portion of the muscle obtained at biopsy is sent for mtDNA analysis. Patients with mtDNA deletions present as sporadic cases (9).
ENCEPHALOMYOPATHIES
The common mitochondrial encephalopathies include MELAS (myopathy, encephalopathy, lactic acidosis, and stroke-like episodes) and MERRF (myoclonic epilepsy and RRFs). MELAS can include psychomotor retardation, ataxia, cognitive impairment, deafness, diabetes mellitus, and limb weakness. Optic atrophy, dystonia, chorea, peripheral neuropathy, myoclonus, pigmentary retinopathy, spasticity, and progressive external ophthalmoplegia (PEO) have also been reported in MELAS patients since the original description. Of all cases meeting the clinical criteria for MELAS, 80% are positive for an A-to-G transition at base pair 3243 within the tRNA LeuUUR gene (10–12). A large number of different mutations have also been found to result in the MELAS phenotype. A G13513A mutation in the ND5 gene was also associated with recurrent focal cortical brain hematomas in a child with MELAS (13). Conversely, of all cases with the 3243 mutation from an unselected population of 17 patients with mitochondrial myopathy, 50% were found to fulfill the criteria for MELAS at the time of clinical assessment (10). Theoretically, further cases could develop these clinical features with the passage of time.
The core clinical features of MERRF are myoclonus, ataxia, and seizures. Myoclonus is often the presenting symptom, and may be induced by action, noise, or photic stimulation. Seizure types are variable, but include drop attacks, focal seizures, and photosensitive tonic–clonic seizures (14,15). Myopathy is usually either mild, subclinical, or absent. RRFs are seen on muscle biopsy in the majority of patients, but not in all cases. Additional associated features have been reported in varying numbers of patients. These include ophthalmoplegia, ptosis, deafness, peripheral neuropathy, headache, foot deformity, optic atrophy, and cervical lipomas. An insidiously progressive dementia may occur later in the disease course. Furthermore, the vast majority of the clinical features seen in the other mitochondrial encephalomyopathies have been reported in patients with MERRF. These include short stature, involuntary movements, episodic hemicranial headache, cardiomyopathy, spastic paraparesis, and psychomotor retardation (2,15–17).
The most commonly detected mutation, found in approximately 80% of cases fulfilling the clinical criteria for MERRF, is at position 8344 within the tRNA Lys, and was first reported by Shoffner in 1990 (10,18,19). It was detected in three independent MERRF pedigrees, was absent in 75 controls, alters a highly conserved nucleotide, and is heteroplasmic, thus fulfilling the criteria for a pathogenic mutation as opposed to a silent polymorphism. In vitro studies using cybrids have confirmed its pathogenic role (20). Other mutations within the same tRNA gene (T8356C (17) and G8363C (21)) are also described in association with MERRF. Insertion of a C nucleotide at position 7472 in the tRNA Serine UCN gene results in a syndrome of hearing loss, ataxia, and myoclonus that is very similar to MERRF (22). MERRF has also been reported in patients harboring multiple mtDNA deletions, (23) and overlap syndromes with features of both MERRF and MELAS have been reported for the T7572C and T8356C tRNA Serine UCN mutations. Retinopathy has only been found in MERRF cases with mtDNA mutations other than the most common A8344G mutation (2,10,24).
NEUROGENIC WEAKNESS ATAXIA AND RETINAL PIGMENTATION
The key features of this mitochondrial encephalomyopathy are peripheral neuropathy, ataxia, retinitis pigmentosa, seizures, and dementia (25). A spectrum of neurologic findings have been described in neurogenic weakness ataxia and retinal pigmentation (NARP), including migraine and mental retardation (25–31). Clinical features are variable as is the age of onset, which in one series varied between 1 and 32 years (32,33). Inheritance is maternal.
The commonest mutation is a T-to-G transversion at nucleotide position 8993. This causes a change from the highly conserved leucine to arginine within subunit 6 of the mitochondrial F0F1 ATP synthase. Patients with NARP usually have above 80% mutant mtDNA levels. With mutant mtDNA levels below 75%, patients usually suffer from pigmentary retinopathy alone, or suffer migraines, or are asymptomatic (25,30). This illustrates the good correlation between mutant load and disease severity that is not present in the majority of the other mitochondrial encephalomyopathies.
RRFs and other morphologic mitochondrial hallmarks are lacking in muscle biopsies from patients with NARP (25) There is, therefore, an overlap between NARP and some nonmitochondrial neurodegenerative disorders such as Refsum’s disease and Usher’s syndrome, which share the clinical features of hearing loss, ataxia, and visual impairment due to retinitis pigmentosa. Lactic acidosis may also be absent in NARP. Muscle weakness is often mild and may be masked by the presence of ataxia. Reduced complex V activity has been found in cultured cells from affected patients (26,34) but no in vivo evidence of ATPase deficiency has been reported in patients carrying the NARP mutation. Biochemical analysis of muscle samples may fail to reveal a respiratory chain or OXPHOS defect however, since complex V (ATP synthase) activity is rarely measured routinely.
LEIGH’S SYNDROME
This subacute necrotizing encephalomyelopathy is characterized by bilateral symmetrical focal necrotic lesions within the thalamus, extending into the pons, inferior olives, and spinal cord. The clinical features of Leigh’s syndrome (LS) are of psychomotor retardation, hypotonia, failure to thrive, respiratory abnormalities, oculomotor disturbances, optic atrophy, seizures, and lactic acidosis. Biochemical abnormalities include defects of oxidative phosphorylation (35) (in particular complex I (36) or complex IV, (37,38) and deficiency of the pyruvate dehydrogenase complex [PDH], (39) and biotinidase deficiency) (40). The majority of LS cases are believed to result from nuclear gene defects (34,41). This has been confirmed for cases of LS with PDH complex deficiency, (41) complex I, (42) and I1 (43). Complex IV–deficient LS results from mutations of the Surf or other assembly genes (44–46). Nuclear gene defects are also presumed to be causal for cases of LS with biotinidase deficiency. Up to 20% of LS patients have the T-to-G, or T-to-C, mtDNA mutation at position 8993 within the ATPase 6 gene of complex V (47–51). Mutant loads are above 90%, and lower levels of this mutation are associated with the NARP syndrome. High levels of the A3243G MELAS mutation and the A8344G MERRF mutation have also been reported in LS (52–54). Other mtDNA mutations described include G1644T within the tRNA Val gene, (55) and deletions (53,56,57) and depletion of mtDNA levels (58). The ATPase 6 mutations T8851C and T9176C are also associated with bilateral striatal necrosis and with maternally inherited LS (14,59,60).
LEBER HEREDITARY OPTIC NEUROPATHY
LHON is characterized clinically by bilateral sequential acute or subacute visual failure due to degeneration of the retinal ganglion cells and their axons. Three mtDNA mutations, G11778A, G3460A, and T14484C, located in the MTND4, MTND1, and nMTND6 genes, respectively, account for approximately 95% of cases. The G11778A mutation is the most common being present in 56% of cases, the G3460A in 31%, and the T14484C in 6.3% (61). The disease occurs predominantly in young men, with usually little or no visual recovery, although the T14484C mutation is generally associated with better prognosis. A range of “secondary” or “intermediate” polymorphisms may modify expression (62). More recently, additional mutations in MTND1 and MTND6 have been described (63,64), with the C4171A mutation in MTND1 being associated with significant visual recovery. All these LHON mutations are all usually present in homoplasmic or high heteroplasmic proportions. Additional features include dystonia and striatal degeneration as reported in some families, especially with point mutations G14459A, A11696G, and T14596A in the mitochondrial complex I subunit genes (65,66).
NUCLEAR GENE MUTATIONS AND MITOCHONDRIAL MOVEMENT DISORDERS
MTDNA POLG MUTATIONS
MtDNA POLG is a heterodimer comprising a 140-kDa α-subunit and a 41-kDa β-subunit. It is located within the inner mitochondrial membrane and is essential for mtDNA replication. The α-subunit is catalytic and contains both polymerase and exonuclease activities, the β-subunit facilitates DNA binding and promotes DNA synthesis (67). Mutations of POLG have been associated with a range of clinical phenotypes, including PD. Autosomal dominant or recessive inheritance of CPEO with age of onset ranging from 10 to 54 years was followed some years later (range 6–40 years) by the development of an asymmetric, levodopa (L-dopa)-responsive bradykinetic rigid syndrome together with resting tremor in some patients. Additional features included variable limb, pharyngeal, or facial weakness, cataracts, ataxia, peripheral neuropathy, and premature ovarian failure (68). Muscle biopsy demonstrated ragged red, COX-negative fibers in all patients with multiple mtDNA deletions on Southern blotting. Symmetrically reduced striatal [18-F]β-CFT was seen in two patients. Brain histology was available on a further two patients, both showed severe loss of substantia nigral dopaminergic neurones but without the development of Lewy bodies or other synuclein aggregates. Four families had the same A2864G mutation inherited in autosomal fashion in three and with a founder effect in the fourth. Mutations in the exonuclease or polymerase portions of the gene were identified in the autosomal recessive families. A further patient with autosomal dominant PEO–parkinsonism and an A2492G mutation has been reported (69).
FRIEDREICH’S ATAXIA
Friedreich’s ataxia (FRDA) is an autosomal recessive disease characterized clinically by a progressive gait and limb ataxia, absence of deep tendon reflexes, and loss of position and vibration sense in the lower limbs. Skeletal abnormalities, cardiac hypertrophy, and to a lesser degree diabetes and optic atrophy are also present in FRDA patients.
FRDA is inherited in an autosomal recessive pattern with over 95% of patients having a homozygous expansion of a GAA triplet repeat (6-34 GAA repeats expanded to between 67 and 1,700 in patients) in intron 1 of the FRDA gene on chromosome 9 (70). Most of the remaining patients are compound heterozygotes with the GAA expansion in one allele and point mutations in the other (71).
The size of the GAA repeat appears to influence the clinical phenotype with a significant inverse relationship between the size of the smaller GAA repeat and the age of onset (72,73). Homozygous GAA repeat expansions resulted in decreased frataxin mRNA levels in patients’ lymphoblasts and fibroblasts (74,75). This correlated with lower levels of frataxin protein in skeletal muscle, cerebellum, and cerebral cortex from patients with FRDA. In addition, frataxin protein levels have been shown to be inversely decreased in proportion to the GAA repeat size in FRDA lymphoblasts (76).
While the normal function of frataxin is not known, the analysis of the protein sequence identified a predicted N-terminal mitochondrial targeting sequence (76) and a stretch of highly conserved amino acids in the C terminal half of the protein (77) Evidence from the knockout of the frataxin homologue in yeast cells, mice models, and patient samples suggests that frataxin deficiency results in impaired mitochondrial respiratory chain function, mitochondrial iron accumulation, decreased mtDNA levels, and an increased oxidative stress.
Dysfunction of the mitochondrial respiratory chain was observed in yeast YFH1 mutants (78,79), conditional frataxin knockout transgenic mice (80), and postmortem heart and skeletal muscle from FRDA patients (81,82). The pattern of respiratory chain dysfunction (complexes I, II, and III) is reminiscent of that caused by oxidative stress caused by knockout of the manganese superoxide dismutase (SOD) (83), or secondary to excitotoxicity in Huntington’s disease (HD) (84). However, all the activities decreased in FRDA contain Fe–S clusters, which led to the proposal that an abnormality of Fe–S cluster synthesis may be responsible for these defects. As the decrease in activities preceded the iron accumulation in the conditional frataxin knockout transgenic mouse model of FRDA (80), the likely mechanism involves decreased Fe–S synthesis, which may then be exacerbated in the longer term by secondary oxidative damage. The concurrent loss of mtDNA (81,85) may also contribute to the mitochondrial respiratory chain (MRC) defects, which may be secondary to increased oxidative damage.
31Phosphorous magnetic resonance spectroscopy (31P MRS) is an in vivo technique that can measure high-energy phosphorous compounds (phosphocreatine [PCr] and ATP) in heart and skeletal muscle. In skeletal muscle, PCr levels fall during exercise, and the analysis of the rate at which PCr levels recover following exercise (Vmax) is a measure of the efficiency of oxidative phosphorylation (86). The PCr/ATP ratio is used as a good measure of energy availability in heart (87). 31P MRS analysis of FRDA patients revealed markedly decreased oxidative phosphorylation in the heart (88) and skeletal muscle, with the latter correlating with the size of the smallest GAA repeat (89). These data underline the role of mitochondrial dysfunction in FRDA and suggest it is playing a primary role in disease pathogenesis.
A conditional knockout mouse model of muscle frataxin leads to death at 10 to 12 weeks (80,90). Abnormalities began at week 4 with a 50% reduction in complex II activity; cardiac function remained normal at this time. By week 5, however, there was an increase in left-ventricular mass, left-ventricular end systolic and diastolic diameters, and a 24% reduction in the shortening fraction. At week 6, cardiac output was down by 15%, and there was evidence of abnormal mitochondrial morphology. Week 7 showed a 67% decrease in shortening fraction and a surprising reduction in oxidized protein. By week 8, cardiac output was 67% of control, complex II activity was down 80%, but there was no evidence of mitochondrial iron accumulation. At week 9, mitochondrial iron was elevated, but lipid peroxidation was decreased. Idebenone treatment 90 mg/kg/day delayed the onset of cardiac abnormalities by only 1 week and prolonged lifespan by 10%. The therapy had no protective effect on complex II activity.
The potential protective effect of antioxidant treatment in this murine model was assessed by overexpression of Cu–Zn SOD and a Mn SOD mimetic. However, there was no evidence of a protective effect on the development or severity of the cardiomyopathy, and no benefit on survival (91). Mn SOD protein expression fell by approximately 50% compared to controls in the heart at 10 weeks. There was no increase in oxidized protein levels in either heart or cerebellum. These results indicate that frataxin deficiency does not lead to oxidative stress, and alternative mechanisms may be involved in mediating the neurodegeneration associated with deficiencies of iron sulphur proteins.
Vitamin E is a naturally occurring lipid soluble antioxidant distributed throughout cellular membranes and is particularly abundant in mitochondrial membranes. Vitamin E treatment has been shown to increase vitamin E levels in a variety of tissues, including brain, muscle, and heart (92). It has been used to treat cardiovascular disease, PD, cancers, and ataxia with vitamin E deficiency (AVED) with varying degrees of success (93–96). The treatment of FRDA patients with vitamin E has only been reported in conjunction with coenzyme Q10 (CoQ10). CoQ10 is naturally found in cells, and up to 5 mg/day may be consumed in an average diet (very rich in soybean oil, meat, and fish). CoQ10 is readily taken up into the blood (97), the brain (98), and liver (92), although other reports suggest dietary CoQ10 levels do not influence tissue CoQ10 levels in the rat (99). CoQ10 may reduce vitamin E, and therefore when combined in a therapy may act synergistically (100). We have assessed the efficacy of long-term treatment of 10 patients with FRDA with high doses of vitamin E (2,100 IU/day) and CoQ10 (400 mg/day). After 6 months, 31P MRS data indicated that heart and skeletal muscle energetics were significantly improved (101). Four-year follow-up data from this study showed the enhanced energy levels were maintained, clinical parameters were stabilized or improved in 7 out of 10 patients, and heart fraction shortening had improved (102). A combination of CoQ10 and vitamin E improved neurologic progression, and low dose was as effect as high-dose therapy in those with coexisting CoQ10 deficiency (103).
Idebenone is a short-chain analog of CoQ10, is well tolerated by humans, crosses the blood–brain barrier (104), has been reported to be a relatively good antioxidant (105), and has been used in a variety of diseases with some benefits (106,107). The effect of idebenone upon cardiac hypertrophy in FRDA patients was assessed using echocardiography, but other clinical improvements were not reported. After 6 months’ treatment, cardiac hypertrophy was decreased in up to half the patients tested, although this was not always associated with improved fraction shortening (108). Another assessment of idebenone failed to identify improvements in skeletal muscle 31P MRS or echocardiographic parameters (109), although this may reflect the short time scale used.
An alternative therapeutic strategy is to use histone deacetylase inhibitors to increase frataxin mRNA levels (110). A preliminary study using high-dose oral nicotinamide in FRDA patients showed increased levels of frataxin in white blood cells over 8-week duration of treatment (111).
COQ10 DEFICIENCY
CoQ10 is a lipophilic component of the respiratory chain which transfers electrons from complexes I and II, and from fatty acids and branched chain amino acids via flavin-linked dehydrogenases, to complex III (Fig. 26.2). The first report of human disease associated with CoQ10 deficiency was in a patient with encephalomyopathy and recurrent myoglobinuria with RRFs and changes of lipid storage on muscle biopsy (112). Relatively severe CoQ10 deficiency was then described in six patients with early-onset (age range birth–16 years) myopathy and ataxia (113). Seizures, weakness, and mental retardation was described in some and cerebellar atrophy found in all. Genetic testing for FRDA and spinocerebellar ataxia was negative; inheritance was consistent with an autosomal recessive pattern. Muscle biopsy in these patients showed only nonspecific abnormalities and, in particular no evidence of mitochondrial pathology. Residual muscle CoQ10 levels were 26% to 35% of normal. Administration of CoQ10 (300–3,000 mg/day) resulted in significant improvement in the ataxia.
Subsequent assay of muscle CoQ10 levels in 135 patients with genetically undefined childhood-onset ataxia identified significantly reduced levels in 10% (114). All patients had cerebellar atrophy, and some had seizures, developmental delay, and pyramidal features. Lactic acidosis in the ataxic patients is rare, and in contrast to the myopathic form, the muscle biopsy may appear normal. The same group subsequently described muscle CoQ10 deficiency in two brothers with adult-onset (aged 29 and 39 years) progressive cerebellar ataxia with cerebellar atrophy and hypergonadotrophic hypogonadism. Muscle morphology showed only neurogenic changes. CoQ10 750 to 1,200 mg/day resulted in improved ataxia, neurophysiology, and normal testosterone levels within 2 months in these patients.
PARKINSON’S DISEASE
The first direct association between mitochondrial dysfunction and idiopathic PD was made with the discovery in 1989 of a complex I defect in substantia nigra from patients with PD (115,116). This study has been expanded over the years, and results to date show that there is a specific circa 35% complex I deficiency in PD nigra. This defect in complex I activity does not appear to affect any other part of the respiratory chain. In addition, no defect in mitochondrial activity has been identifiable in any other part of PD brain, including caudate putamen, globus pallidus, tegmentum, cortex, cerebellum, or substantia innominata (117).
This observation provided a direct biochemical link between idiopathic PD and the 1-methyl 4-phenyl 1,2,3,6 tetrahydropyridine (MPTP) model of this disorder. 1-methyl 4-phenylpyridinium (MPP+), the toxic metabolite of MPTP, inhibits complex I in a concentration-dependent but reversible manner. Prolonged exposure of submitochondrial particles to MPP+, however, results in more severe and irreversible inhibition of complex I when electron flow through the chain is prevented by inhibition of COX (complex IV). This severe and irreversible inhibition can be prevented with free-radical scavengers (118). Free radicals can damage the respiratory chain. In vitro studies suggest that both complexes I and III are particularly affected, while in vivo studies suggest that complex IV and, less so, complex I are inhibited (119–122).
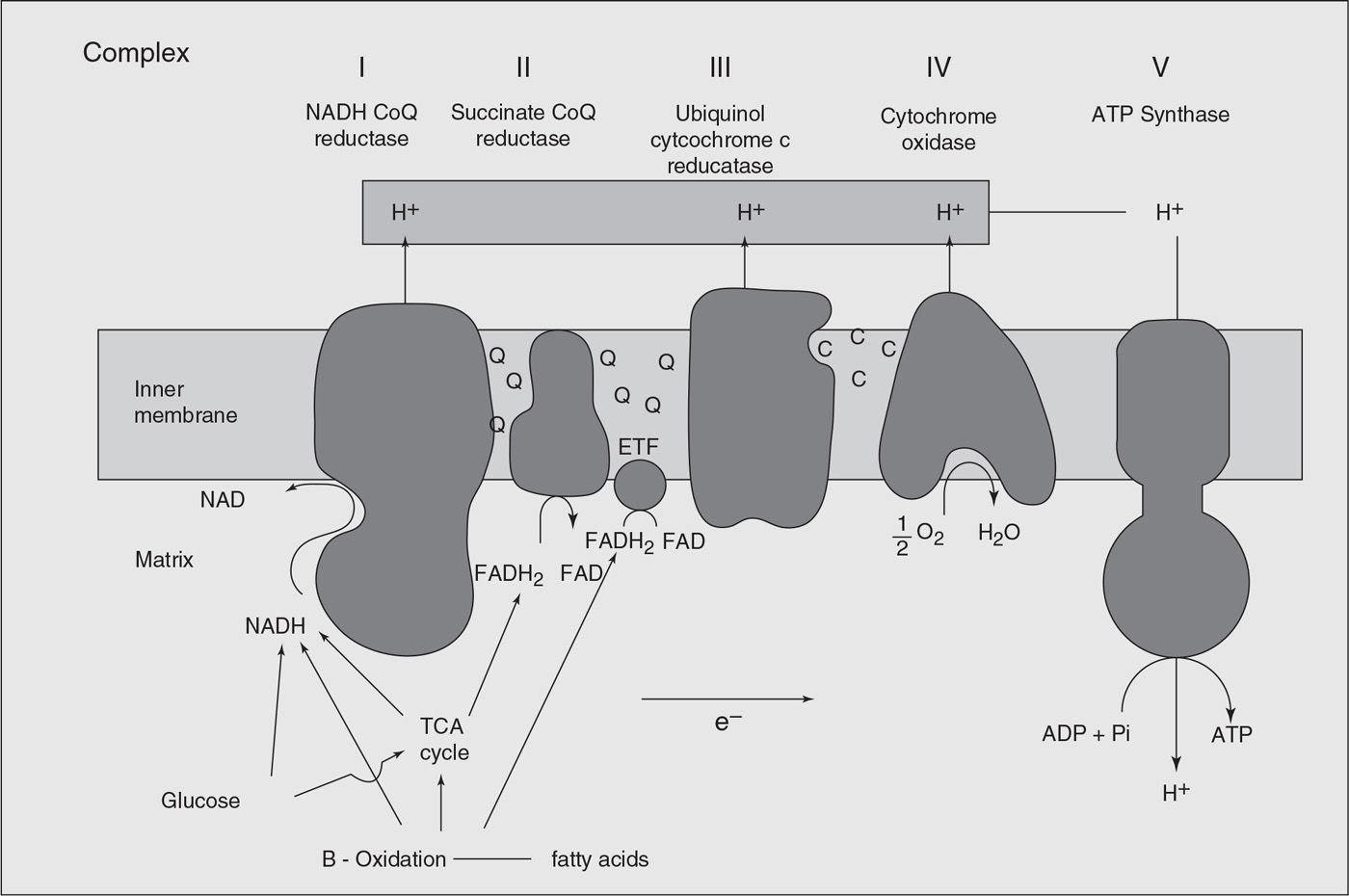
Figure 26.2. The functional features of the mitochondrial respiratory chain in oxidative phosphorylation system. ADP, adenosine diphosphate; ATP, adenosine triphosphate; CoQ, ubiquinone; e−, electron; ETF, electron transfer flavoprotein; FAD, flavin adenine dinucleotide; FADH2, the reduced form of flavin adenine dinucleotide; NAD, nicotinamide adenine dinucleotide; NADH, the reduced form of nicotinamide–adenine dinucleotide; Pi, inorganic phosphate; TCA, tricarboxylic acid cycle.
Following the report of complex I deficiency in PD substantia nigra, respiratory chain abnormalities were described in skeletal muscle mitochondria from PD patients. This particular area has proved very contentious, with several groups describing either similar defects or no abnormality whatsoever (123). Two magnetic resonance spectroscopy studies on skeletal muscle mitochondrial function in PD have shown conflicting results (124,125).
Finally, mitochondrial complex I deficiency was also identified in platelet mitochondria of PD patients (126). In contrast to skeletal muscle, there is a consensus amongst several laboratories that complex I deficiency does exist in PD platelet mitochondria. The majority of studies, however, suggest that this deficiency, as least based on a group-to-group analysis, is modest (circa 20%–25%) (123).
The cause of the complex I defect in PD remains unknown. As virtually all the brains taken from PD patients had been exposed to L-dopa, an important question related to whether L-dopa caused the complex I deficiency. There was no relationship between the level of complex I defect and prior L-dopa exposure. Furthermore, there is no deficiency of complex I activity in PD striatum, an area high in dopamine (127). L–Dopa does not appear to cause any deficiency of complex I activity in platelet mitochondria, which again might be expected given the relatively high circulating levels of L-dopa in the periphery. Most importantly, it has been shown that patients with MSA who have taken L-dopa in quantities and for duration comparable to patients with PD have no defect of mitochondrial activity in their substantia nigra (128). Also, cell loss in the nigra is at least as severe in MSA as it is in PD, and so one would expect that, if the mitochondrial defect in PD were simply a reflection of this degeneration, the same abnormality should be present in MSA. Its absence in MSA therefore suggests that its presence in PD is the result of a more-specific cause than simple cell loss.
Alternatives for the cause of the complex I deficiency in PD include an mtDNA mutation or nuclear gene mutations of mitochondrial proteins. Following the identification of the complex I defect in PD, several studies investigated the structure of mtDNA in tissues from PD patients. An early study suggested increased levels of the common deletion in PD brain (129). However, this study did not use age-matched controls, and the increase in the common deletion was in fact no greater than would be expected from the aging process. Other studies using properly matched controls found no increase in this mitochondrial mutation in PD substantia nigra (130,131). Several studies have sequenced mtDNA in PD, but these have all involved small numbers of unselected patients (132–139). While some reports have suggested an increased frequency of certain mtDNA polymorphisms in PD, this has not been replicated in other studies (140,141).
The relationship between mitochondrial dysfunction, mtDNA, and PD has been highlighted further by the description of mtDNA abnormalities in PD patients. Occasional mtDNA point mutations have been identified in PD, but these have not been present in the general PD population. Thus, their association with PD may merely represent part of the wide clinical spectrum of mtDNA mutations and not necessarily imply a more common role in sporadic PD. POLG mutations have been described in patients with CPEO and PD with RRFs and multiple mtDNA deletions, but mutations of this gene have not been found in sporadic PD (142). A mutation in the mtDNA 12S RNA was found in a patient with maternally inherited early-onset PD, deafness, and neuropathy (143). Several studies have sequenced mtDNA in PD patients, and no consistent mutation has been described (144). However, none of the studies to date has focused on PD patients with complex I deficiency. Two studies have demonstrated a relationship between mtDNA haplotypes and the risk for developing PD. The first showed a reduced risk for PD in individuals with haplotypes J and K (145), and the second a 22% decrease in PD in those with the UKJT haplotype cluster (146). In contrast, a smaller study reported an increased risk for PD with haplotypes J and T (147).
Two studies have used genetic transplantation with calls lacking mtDNA (rho zero cells) to investigate the potential for PD mtDNA to determine the complex I defect. In one, unselected PD patients’ cells were fused and grown in mixed culture (148). In another, PD patients were selected on the basis of demonstrating a peripheral complex I deficiency. These patients’ cells were then fused with rho zero cells and grown both in mixed and clonal culture (149
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


