Childhood movement disorders are both similar and distinct from movement disorders in adulthood. While the two overlap in their definitions of specific movements, this chapter will serve to highlight the many differences encountered. In this chapter, the classification of childhood movement disorders is divided into four major areas: transient developmental, paroxysmal, noninherited secondary, and hereditary and metabolic. To avoid overlap, selected movement disorders such as Tourette syndrome (TS) (Chapter 30), dystonia (Chapter 28), and chorea (Chapter 20) will be covered in other chapters.
GENERAL APPROACH TO THE CHILD WITH A MOVEMENT DISORDER
Movement disorders require observation by a knowledgeable physician. Since children with paroxysmal disorders do not often have attacks while in a physician’s office, video documentation from the family is essential. From the history, one should determine whether the problem is acute or chronic, paroxysmal or continuous, static or associated with the loss of previously acquired skills, or whether there is evidence for multiple system involvement. The description of the symptoms should include age of onset, type of movement, progression, focality, timing, triggers, patient’s ability to control, presence during sleep, effect on activities, associated difficulties, and other observations. Historical details about the patient’s gestation, delivery, early development, previous illnesses, drug history, exposures, and social and family histories are often helpful for proper classification. Clues for separating paroxysmal dyskinetic disorders from epilepsy include the identification of specific movement abnormalities (for example dystonia, choreoathetosis, tics, or stereotypies), maintenance of consciousness, ability to stop with distraction, a normal respiratory pattern, lack of a postictal state, and absence of an electroencephalographic (EEG) abnormality during attacks. A comprehensive, general examination is essential for properly defining the movement and for identifying clues that indicate a systemic problem.
TRANSIENT DEVELOPMENTAL DISORDERS
Transient developmental disorders typically occur in otherwise-healthy infants or young children with no evidence of structural brain abnormalities or metabolic etiologies. They are characterized by a pattern of abnormal movements, a lack of progression, minimal disability, absence of specific laboratory markers, typical development, normal neurologic function, and complete resolution. Since they may be manifestations of a developing neural system, labeling them as a disorder is controversial. There is little role for pharmacotherapy, and treatment should focus on parent education (Table 39.1).
| Transient Developmental Disorders and Their Differential Diagnoses |
Symptom | Differential Diagnoses |
Jitteriness | Primary jitteriness Secondary jitteriness: maternal selective serotonin reuptake inhibitor (SSRI) use or drug withdrawal, metabolic derangement, hypoxic ischemic encephalopathy (HIE) |
Dystonia | Physiologic Fever-induced Benign idiopathic dystonia |
Myoclonus | Benign neonatal myoclonus Benign myoclonus of early infancy Neonatal seizures, infantile spasms Hyperekplexia Opsoclonus myoclonus |
Shuddering | Primary shuddering attacks “Wet dog shakes” Primary jitteriness Drug induced Benign myoclonus of early infancy |
Torticollis | Benign paroxysmal torticollis Sandifer syndrome Spasmus nutans Cervical dystonia Ocular torticollis Positional torticollis/plagiocephaly Drug induced Infection/inflammatory Increased intracranial pressure (ICP) |
Head nodding | Periodic head nodding Spasmus nutans Bobble-head doll syndrome Stereotypy Sandifer syndrome Rhombencephalosynapsis |
CHOREA/DYSTONIA
Physiologic
Many newborns and infants manifest a variety of dyskinetic movements that tend to be brief in duration and of no apparent pathologic consequence. Pursing and sucking of the lips, head and neck extensions, body and extremity twists, turns and postures are but a few of the myriad of movements labeled as “physiologic chorea” or “dystonia.”
Fever-Induced Dystonia
Two children (aged 2.5 and 6 years) have been reported to have a syndrome of fever-induced dystonia. Episodes consisted of lower-extremity dystonia with foot inversion and great toe dorsiflexion. Episodes diminished with age. Metabolic and imaging studies were normal. Each child had a similarly affected parent (1).
Benign Idiopathic Dystonia of Infancy
Several normal children have been described with transient dystonic postures, usually shoulder abduction, forearm pronation, and wrist flexion that appear in the first months of life, briefly progress, and then resolve after 3 months to 5 years. Posturing of the upper extremity, body, or trunk can be intermittent or persistent, occur at rest, and disappear with volitional movements. Neurologic examination is otherwise normal, and developmental outcome is good (2,3). This disorder can sometimes be mistaken for orthopedic disorders, brachial plexus injury, or hemiplegic cerebral palsy (CP) (4).
MYOCLONUS
Benign Neonatal Myoclonus
Benign neonatal myoclonus is characterized by neonatal onset and myoclonic jerks that are repetitive, can be symmetric, and occur in clusters lasting for several minutes before stopping spontaneously. Myoclonic movements are largely confined to sleep and appear most frequently during quiet stages, less often during rapid eye movement (REM) sleep. Myoclonic movements typically resolve by 2 to 4 months of age, although up to a third may persist beyond 3 months. Nearly all patients have resolution by 1 year of age (5). The presentation of benign neonatal myoclonus can be concerning for epilepsy; however, EEG is normal. Familial cases have been reported and can follow an autosomal dominant pattern. Benign neonatal myoclonus has been shown not be linked to genetic mutations associated with benign familial neonatal seizures, that is, KCNQ2 or KCNQ3 mutations (6).
Benign Myoclonus of Early Infancy (Fejerman Syndrome)
Benign myoclonus of early infancy is frequently misdiagnosed as infantile spasms (7); however, EEGs are normal. Myoclonus can occur several times a day, is present during wakefulness and sleep, but tends to be more apparent when feeding or playing. In addition to myoclonus, children may also have the coexistence of clusters of paroxysmal nonepileptic motor phenomenon in muscles of the neck, upper limbs, and trunk, including tonic spasms, atonia or negative myoclonus, shuddering, or a combination of different movements. Episodes typically begin between 3 and 9 months of age, and resolve by 6 to 30 months of age. Outcomes appear to be favorable, without long-term cognitive issues (8).
JITTERINESS
Neonatal jitteriness, also called neonatal hyperexcitability, is characterized by a low-amplitude and high-frequency tremor that involves the chin and extremities. Movements tend to be stimulus-sensitive and are exacerbated with crying or startle (9). The etiology of jitteriness may not be known, but secondary causes can include drug withdrawal, metabolic derangements, and hypoxic ischemic encephalopathy. More than one-third of selective serotonin reuptake inhibitor (SSRI)-exposed infants had clinical signs including jitteriness, but most were mild and self-resolved (10). In children without risk factors for secondary jitteriness, the time course of symptoms varied, with three-quarters having improvement by 9 months of age and all resolving by 2 years (11).
SHUDDERING ATTACKS
Shuddering or shivering attacks are a benign movement disorder characterized by periods of rapid shivering of the head, shoulder, or arms. Attacks are brief and can occur up to hundreds of times per day with no associated loss of awareness. Episodes start in infancy or early childhood and resolve within the first decade. Based on a strong family history of essential tremor, it has been hypothesized that shuddering attacks may be a precursor for essential tremor (12), although this is controversial (13). One patient has been successfully treated with β-blockers (14).
TORTICOLLIS
Benign Paroxysmal Torticollis
Benign paroxysmal torticollis typically occurs within the first 12 months of life. Attacks occur without a known specific trigger and consist of a head tilt to either side, sometimes accompanied by irritability, tortipelvis, vomiting, pallor, or vertigo/ataxia (15). Laterality of the torticollis can sometimes alternate. The frequency of attacks varies, and duration can range from minutes to weeks. There is often improvement by the age of 2, and usually resolution of symptoms by age 3 years. It has been hypothesized that vestibular dysfunction may play a role based on abnormal audiometry and calorics (16,17), but in most subsequent studies, testing was normal. There is a strong association with paroxysmal vertigo, positive family history of migraine, and many children go on to develop migraines. It has been suggested that genes implicated in hemiplegic migraine, including CACNA1A and PRRT2, may also be associated with this disorder (18,19). There is no good symptomatic treatment for benign paroxysmal torticollis.
Sandifer Syndrome
Sandifer syndrome is associated with gastroesophageal reflux and may consist of dystonic movements of the head and neck, retrocollis, or sometimes posturing of the body or opisthotonos. Repetitive side-to-side head movements have also been described in older children (20). Episodes are typically associated with feedings or may occur postprandial. Diagnosis is made with pH probe to document reflux. Symptoms often resolve with gastrointestinal intervention (21). The pathophysiologic mechanism of Sandifer syndrome remains unclear.
Spasmus Nutans
Spasmus nutans is characterized by the triad of head nodding (horizontal or vertical), rapid asymmetric low-amplitude nystagmus (monocular or binocular), and torticollis. It typically appears between 3 and 12 months of age and resolves by 3 to 5 years of age. Low socioeconomic status may be a risk factor (22). There have been associations with multiple ocular abnormalities such as optic nerve pallor, refractive error, strabismus, rod/cone dystrophies, and intracranial disorders such as optic nerve hypoplasia, optic nerve and chiasmal gliomas, and Chiari I malformations (23). Electroretinogram and neuroimaging should be obtained as part of the evaluation of spasmus nutans.
HEAD NODDING
Periodic head nodding, or repetitive flexion of the neck, can occur in vertical, horizontal, or oblique planes and are often seen in otherwise-normal children. Movements can spontaneously resolve in several months, or persist (see motor stereotypies). Head nodding has also been reported in children with a family history of essential tremor, infantile esotropia, and rhomboencephalosynapsis (24). Overall, developmental outcomes are good. Head nodding should be differentiated from the more serious “bobble-head doll” syndrome, in which children have jerky head movements at 2 to 3 Hz, resembling a doll’s head on a spring. Bobble-head doll syndrome is usually found in conjunction with an intracranial abnormality involving expansion of the third ventricle via third ventricle cyst, suprasellar arachnoid cyst, or aqueductal stenosis. Symptoms usually resolve with surgical intervention, and prognosis improves with earlier diagnosis (25).
PAROXYSMAL TONIC UPGAZE OF INFANCY
This disorder is characterized by repeated episodes of tonic sustained upward deviation of the eyes with compensatory neck flexion lasting hours to days. Horizontal eye movements are normal, but downbeating saccades are often present with attempted downgaze. The episodes are exacerbated by fever or illness and improve with sleep. At times, symptoms are accompanied by ataxia, but often an otherwise-normal neurologic exam. There have been associations with hypomyelination, periventricular leukomalacia (PVL), and vein of Galen malformations, but often neuroimaging is normal. Half of cases have a good outcome, while the other half may have residual ataxia, cognitive difficulties, or oculomotor abnormalities. Improvement with levodopa (L-dopa) treatment has been reported (26).
PAROXYSMAL MOVEMENT DISORDERS
Paroxysmal movement disorders are the most common movement abnormalities encountered by child neurologists. The list of nonepileptic paroxysmal movement disorders is extensive, and it is often helpful to separate the various conditions on the basis of the type of movement disorder observed (Table 39.2). Establishing a clear understanding of the abnormal movement frequently requires viewing a video of the child’s attack, as history alone may be vague or inconclusive. After fully assessing the child, the physician in conjunction with the patient, the family, and school personnel can determine whether the episodic movements are having a negative functional or psychological impact. Medications should be reserved for problems that are disabling and not easily remedied by other interventions.
ATAXIA
Several familial forms of episodic ataxia have been identified, each representing a different genetic disorder (see Chapter 35).
| Paroxysmal Movement Disorders |
Symptom | Differential diagnoses |
Ataxia | Episodic ataxia without myokymia Episodic ataxia with myokymia Episodic ataxia with paroxysmal choreoathetosis Paroxysmal tonic upgaze with ataxia Familial periodic ataxias |
Dyskinesia | Paroxysmal dystonic choreoathetosis Paroxysmal hypnogenic dyskinesia Secondary paroxysmal dyskinesia Paroxysmal kinesigenic dyskinesia Paroxysmal nonkinesigenic dyskinesia Intermediate or exertional dyskinesia |
Tics | Provisional tics Chronic motor or vocal tic disorder Tourette syndrome Secondary tics |
Stereotypy | Primary: with typical development Secondary: autism, Rett syndrome |
Startle | Hyperekplexia Startle epilepsy Brain stem reticular reflex myoclonus |
Posturing during masturbation |
|
Restless legs |
|
Chin quivering |
|
CHOREOATHETOSIS OR DYSTONIA
A commonly used classification identifies four variants (see Chapter 37): paroxysmal kinesigenic dyskinesia (PKD), paroxysmal nonkinesigenic dyskinesia (PNKD), paroxysmal intermediate or exertional dyskinesia (PED), and paroxysmal hypnogenic dyskinesia (PHD) (27). PKD has been associated with mutations in the PRRT2 gene on chromosome 16; it often includes familial infantile convulsions and paroxysmal choreoathetosis (28). PKND has been associated with mutations in the MR-1 gene on chromosome 2 (29). Secondary paroxysmal dyskinesias have been reported in association with calcium or other metabolic derangements, thyroid dysfunction, CP, head trauma, medullary hemorrhage, and spinal cord lesions (30). An additional secondary cause of paroxysmal exertion-induced dyskinesia, glucose transporter 1(GLUT1), is discussed below.
GLUT1 DEFICIENCY
GLUT1, encoded by SLC2A1, is responsible for transport of glucose into the brain (31). Children with GLUT1 deficiency syndrome typically present with seizures early in life, cognitive problems, and developmental delay. A lumbar puncture with low glucose can be diagnostic. The clinical picture of GLUT1 deficiency has expanded and now includes multiple movement disorders such as ataxia, chorea, dystonia, and myoclonus (32). Additional paroxysmal events, such as PED, may also occur (31). Movement abnormalities can occur in the absence of seizures but may still be responsive to the ketogenic diet. The latter is the treatment of choice for GLUT1 deficiency because it provides an alternate form of energy that is able to cross the blood–brain barrier (33).
TICS
Tic disorders represent the most common movement disorder seen by physicians caring for children. TS, once considered a rare disorder, now has an estimated prevalence of 1 to 10 per 1,000 children and adolescents. TS is common in children with autistic spectrum disorders, but its presence is unrelated to the severity of autistic symptoms (34).
STEREOTYPIES
Motor stereotypies are rhythmic, repetitive movements that have a predictable pattern and location, seem purposeful but serve no obvious function, tend to be prolonged, and stop with distraction. Common examples of complex motor stereotypies (CMSs) include arm and hand flapping, waving, or wiggling, often accompanied by mouth opening and neck extension. While previously thought to be associated only with autistic spectrum disorders, intellectual disability, or sensory deprivation, CMSs are now recognized to occur as a “primary” form in otherwise typically developing children. Primary CMS usually begins before age 3 years, persists throughout childhood, and diminishes in severity over time. Movements are provoked by excitement, stress, or being engrossed in activities. The typical duration is several seconds, although movements can last for minutes or longer. Common coexisting conditions include attention-deficit/hyperactivity disorder (ADHD), obsessive–compulsive disorder (OCD), and tics. About one-third of affected children performed significantly worse than controls on tests of motor inhibition and speed, consistent with the diagnosis of a motor coordination disorder. Stereotypy severity is significantly associated with inattention and executive dysfunction, but not motor difficulties. With respect to treatment, there is no good pharmacotherapy, and ongoing research continues to show that behavioral therapy is beneficial. A possible genetic inheritance has been suggested in typically developing children based on a family history of motor stereotypies in 25% of first-degree relatives; results of genetic studies are pending.
The underlying pathophysiology for CMS is unknown. Parallel interacting cortical–striatal–thalamo–cortical circuits provide the framework for understanding this movement disorder, especially the habitual behavioral circuit between premotor cortex and putamen. The absence of premotor cortical potentials in primary CMS suggests that the movements do not utilize typical voluntary pathways within the brain. Two small volumetric neuroimaging studies have been performed in children with primary CMS. In the first, involving six participants, reductions were identified in the caudate and frontal white matter (35). In a more recent study of 19 children with primary CMS, significant reductions were identified in right and a trend in the left putamen volume. Additional preliminary results from high-field 1H-MR spectroscopy at 7.0 Tesla have shown a reduction of γ-aminobutyric acid (GABA) in the anterior cingulate cortex and striatum.
POSTURING DURING MASTURBATION
Infantile masturbation (or gratification behavior) is characterized by stereotyped episodes involving posturing of the lower extremities with pressure on the perineum, no change in consciousness, diaphoresis, grunting, cessation with distraction, and otherwise-normal evaluation. Self-stimulation has been mistaken for epilepsy, dystonia, or dyskinesia. Observation can help clarify the diagnosis and eliminate the need for unnecessary diagnostic tests (36,37).
STARTLE: HYPEREKPLEXIA
Hyperekplexia is characterized by neonatal hypertonia, exaggerated startle response to auditory or tactile stimulation, and apnea. Flexion of the head and limbs toward the trunk can alleviate episodes of an episode, especially in the instance of life-threatening hypertonia and apnea (38). Hyperekplexia is caused by defective glycine transmission across the synapse, and has been associated with postsynaptic glycine-receptor mutations in glycine-receptor subunits GLRA1 (39) and GLRB (40) and with the presynaptic glycine transporter gene SLC6A5 (41). Disruptions in receptor clustering proteins, such as gephyrin, can also be causative. Inheritance is often dominant, but recessive forms exist as well. Phenotype is dependent on the mutation involved, with apnea more common in SLC6A5 mutations and developmental delay more often being found with GLRB mutations (42). Patients respond to treatment with clonazepam (38).
CHIN QUIVERING
Hereditary chin trembling (or hereditary geniospasm) is a rare autosomal dominant condition characterized by paroxysmal movement of the chin and lower lip. Episodes may be triggered by stress or emotion. Onset is in infancy or early childhood, and trembling tends to diminish with age. A linkage to markers on the long arm of chromosome 9 (9q13–21) has been shown (43), but families without this linkage have also been described (44). Botulinum toxin has been used to control the jerks of the mentalis muscle (45).
NONINHERITED SECONDARY CAUSES OF MOVEMENT DISORDERS
Noninherited secondary causes of movement disorders can be due to numerous types of damage or injury to the nervous system (Table 39.3).
In these conditions, the movement abnormalities may occur at the time of the insult, while the patient is recovering from other neurologic deficits, or after a period of neurologic stability. Although a variety of brain insults can subsequently lead to abnormal movements, in infants and children they usually appear after prematurity, birth injury, encephalitis, trauma, and stroke. In some children, in whom the initial brain insult was at age 2 years or before, the onset of a progressive movement disorder (often dystonia) occurred more than 25 years later (46). Many of the etiologies listed in Table 39.3 are common to both pediatric and adult patients, so the focus in this chapter will be on more common pediatric diagnoses such as CP, movement disorders associated with streptococcal infections, and certain postinfectious and autoimmune disorders.
PERINATAL CEREBRAL INJURY: CEREBRAL PALSY
CP is not a single disease entity but rather a broad term used to describe a heterogeneous group of syndromes that cause a nonprogessive disorder arising early in life, of cerebral origin, with abnormal control of movement and posture. The spectrum of motor dysfunction in CP is broad, and despite an underlying static lesion, movement problems may vary over time. The prevalence of CP is 2 to 3.5 per 1,000 live births, with low-birth-weight infants being at higher risk with a prevalence in that population of 90 per 1,000 neonates less than 1,000 g (47).
| SecondaryNoninherited Causes of Movement Disorders in Childhood |
Category | Examples |
Perinatal injury | Hypoxic ischemic injury Perinatal stroke Kernicterus |
Structural lesions | Tumor Trauma |
Vascular | Ischemic stroke Hemorrhagic infarction |
Infectious | Encephalidities (influenza, measles, SSPE, varicella, coxsackie, HIV) |
Autoimmune/inflammatory | Poststreptococcal (PANDAS, Sydenham) ADEM Systemic lupus erythematosus Antiphospholipid syndrome Anti-NMDA-receptor antibodies |
Drug related | Dopaminergic: neuroleptics, metoclopramide, reserpine, l-dopa Anticonvulsants: valproate, phenytoin, vigabatrin Chemotherapy: vincristine, ARA-C, adriamycin Toxins: manganese, CO, cyanide, methanol Psychiatric: SSRIs, lithium, buspirone |
Endocrine | Thyroid dysfunction Addison disease Hypoparathyroidism Pseudohypoparathyroidism |
The mechanisms for CP are extensive, with pre-, peri-, and postnatal causes all possible. Of the prenatal causes, abnormalities that arise during the first trimester are usually related to gross brain malformations such as lissencephaly or schizencephaly. Insults in the second trimester are usually white matter injury, specifically PVL. PVL can be focal or diffuse, and cystic or noncystic. There is likely accompanying damage to deeper structures in addition to cerebral white matter in PVL (48). Third-trimester or perinatal injuries more commonly involve the cortex or deep gray structures, and can include perinatal stroke, hypoxic ischemic encephalopathy, or infection. Postnatal events such as infection, trauma, or stroke can also lead to CP.
Spastic Type
Spastic CP can be further divided into subtypes based on the distribution of impairment, but all of which are associated with hypertonia, hyperreflexia, clonus, and abnormal plantar responses. Spastic hypertonia is defined by the presence of resistance to an externally imposed movement that increases with increasing speed and varies with the direction of joint movement, and has a “catch” that occurs above a threshold velocity. There may be an initial hypotonic phase in infancy prior to development of hypertonia. The child with spastic CP is typically more prone to develop early contractures and have more frequent orthopedic problems than children with dyskinetic CP.
Hemiplegic Subtype
This is the most common subtype of spastic CP with findings localized to one extremity, usually with the upper extremity more involved than the lower. Hemiplegic CP in full-term infants is usually associated with prenatal or perinatal stroke, but can also be secondary to cerebral dysgenesis. Incidence of seizures approaches 70%. If a prior stroke is detected on imaging, then further evaluation for possible hypercoagulable state is recommended (49).
Diplegic Subtype
This is most common in premature infants, and is associated with the presence of PVL. The upper extremities tend to be minimally impaired with lower extremities more impaired.
Quadriplegic Subtype
This is the most severe form, with all four limbs significantly involved and with considerable compromise of motor function. Spastic quadriplegia in the full-term infant may be the result of prenatal insults, brain malformations, or perinatal asphyxia. There may be many accompanying symptoms, including intellectual disability, epilepsy, and microcephaly.
Management of Spastic CP
Pharmacologic treatments can include diazepam, dantrolene, oral baclofen, intrathecal baclofen, and tizanidine, although a recent practice parameter noted insufficient evidence for many of these treatments (50). Selective dorsal rhizotomy may improve gross motor outcomes in some patients with spastic CP (51). Some children may also require orthopedic surgeries such as tendon lengthening or transfers, osteotomies, and joint fusion procedures.
Dyskinetic (Choreoathetoid, Extrapyramidal) Type
Dyskinetic CP is characterized by involuntary movements such as chorea, athetosis, and dystonia. These movements typically begin after age 2 years, and may progress slowly for several years. The movements often persist into adulthood. Abnormal movements usually involve all four extremities, with the upper extremities being more involved than the lower extremities. Dyskinetic forms of CP, especially those associated with athetosis, tend to occur in term infants with perinatal hypoxic ischemic encephalopathy. A smaller number of children with first and second trimester lesions can also have dyskinetic CP (52). It is important to follow the patient for progressive changes and to consider a broader differential, since the etiology for dyskinetic CP may be heterogeneous and include metabolic or genetic components.
Kernicterus, caused by severe untreated hyperbilirubinemia in the newborn period, was previously a frequent cause of dyskinetic CP, but rates of kernicterus have declined. High levels of bilirubin are toxic to the globus pallidus, subthalamic nucleus, cerebellum, and auditory and vestibular pathways.
Ataxic Type
The ataxic or cerebellar form represents a clinically and etiologically heterogeneous group. Associated findings include truncal titubation, dysmetria, and abnormal eye movements. Children with ataxic syndromes usually have a prenatal etiology such as a developmental cerebellar abnormality. Neuroimaging may be normal, or may show infratentorial or biparietal lesions (53).
STREPTOCOCCUS INFECTIONS AND POSTINFECTIOUS DISORDERS
Sydenham’s Chorea
Sydenham’s chorea (SC), the neurologic manifestation of rheumatic fever, is the most common form of immune chorea. Despite its decline in frequency worldwide, it remains the most prevalent form of chorea in children, even in developed areas. The usual age of onset of SC is 5 to 15 years, females > males, but some patients have developed chorea during the third decade of life. Patients typically develop SC 4 to 8 weeks after an episode of Group A β-hemolytic streptococci (GABHS) pharyngitis, and SC is present in about 10% to 20% of patients with rheumatic fever.
SC is defined by the presence of generalized chorea (hemichorea in about 20%–35%). Chorea, however, is often predated by the appearance of neuropsychiatric symptoms, including obsessive–compulsive behaviors (20%–70%), personality changes, emotional lability, distractibility, anxiety, age-regressed behaviors, and anorexia. Other clinical symptoms include motor impersistence (tongue darting, milkmaid, and pronator signs), hypometric saccades, reduced muscle tone, tics, grimacing, clumsiness, dysarthria, and weakness. Cardiac involvement, especially affecting the mitral valve, is present in 60% to 70%, and arthritis is less common (about 30%). Most symptoms resolve in 1 to 6 months, although persistent active chorea has been reported in about 25% for several years. Recurrences occur in about one-third; triggers include GABHS or other infections, oral contraceptive agents, pregnancy, and unknown.
Selected tests should be performed to identify evidence for a streptococcal infection and to identify possible alternative etiologies. Laboratory studies assist in eliminating alternative causes of chorea, but do not confirm the diagnosis of SC. Throat cultures may confirm a preceding GABHS infection, but cultures are positive in only a minority of cases. Acute-phase reactants (erythrocyte sedimentation rate and C-reactive protein) are usually normal in SC, since the interval between the GABHS infection and onset of chorea is relatively long. The percentage of patients with elevated antistreptococcal titers has ranged from about 15% to 30%. Imaging is primarily recommended to rule out other causes of chorea. MRI studies are usually normal in SC, but have been shown to have basal ganglia enlargement and increased T2 intensity during the acute phase. Positron emission tomography and single-photon emission computed tomography have identified transient increases in basal ganglia metabolism. EEG changes are seen in about 30% to 85% of patients, but are nonspecific.
An antineuronal antibody-induced mechanism in SC was initially supported by the finding of IgG reactivity to neuronal cytoplasm in human caudate and subthalamic nuclei that appeared to correlate with the severity and duration of clinical attacks (54). Currently, it is suspected that dopamine (D1 and D2) receptors are the primary antibody target (55,56), although cross-reactive antibodies are also generated, which bind to CNS lysoganglioside-GM 1 (57), and the cytoskeletal protein tubulin (58). Despite the lack of a definitive specific epitope on neuronal cells, the mechanism causing neurologic symptomatology is believed to involve the alteration of neuronal cell signal transduction via CaMKII activation (57). In animal models, rats immunized with GABHS developed antibodies against D1 and D2 receptors and clinically show compulsive-like behaviors (59), and passively-transferred serum obtained from GABHS-immunized mice caused behavioral disturbances (60).
The treatment of patients with SC should include the triad of primary therapy, symptomatic therapy, and supportive care. It is generally recommended that penicillin therapy be given, despite the fact that most patients do not have active GABHS infections. Several studies have suggested that valproate, pimozide, risperidone, haloperidol, tiapride, tetrabenazine, and carbamazepine may be effective for the symptomatic treatment of chorea. SSRIs are effective for obsessive–compulsive behaviors, benzodiazepines are used for anxiety, and in some patients, antidepressants are required. Irrespective of the choice of medication, once the patient has become symptom-free for at least 1 month, consideration should be given to gradually reducing the dosage. Recognizing the autoimmune nature of SC, intravenous immunoglobulin (IVIG), plasma exchange, and oral prednisone have been used in small studies and shown to accelerate recovery. The current recommendation is to reserve autoimmune therapies for patients with refractory chorea, that is, have been unsuccessfully treated with other antichorea medications.
Pediatric Autoimmune Neuropsychiatric Disorder Associated with Streptococcus Infection
PANDAS, modeled upon SC, is proposed to be a GABHS-induced autoimmune disorder. The formal diagnosis requires the affected individual to meet five specific criteria: prepubertal onset, OCD and/or a tic disorder, the dramatic sudden explosive onset of symptoms, a relapsing and remitting course of symptoms that are temporally associated with GABHS infections, and the presence of other neuropsychiatric abnormalities (hyperactivity, emotional lability, anxiety, or piano-playing choreiform movements). Historically, several reports, some published under the heading PITANDS (pediatric infection-triggered autoimmune neuropsychiatric disorders), suggested that a variety of infectious agents could cause acute neuropsychiatric symptoms, including Borrelia burgdorferi, herpes simplex virus, varicella zoster virus, human immunodeficiency virus, Mycoplasma pneumoniae, and the common cold. Nevertheless, because of a suggested link between OCD and SC, the major diagnostic criterion for PANDAS remains its temporal association with GABHS.
PANDAS has been a controversial diagnostic entity based on concerns about its defining features, clinical criterion, proposed temporal association with GABHS, role of other potential infections, inconsistently operationalized criterion, and likely missed diagnoses (61–68). The essence of PANDAS is the temporal association between the onset and exacerbation of tic or OCD symptoms and a GABHS infection. Correlation of the initial episode of symptoms may be difficult, if similar to SC, since the appearance may lag behind the inciting infection by many months. In contrast to the initial event, however, recurrences of symptoms should occur within several weeks of a new infection. Problematic laboratory support has included single-point-in-time assessments of antistreptococcal antibodies, conflicting results as to whether children with OCD, tic disorder, or TS had a streptococcal infection in the 3 months or year prior to the onset of neuropsychiatric symptoms, and findings in two longitudinal studies showing that there was little association between clinical exacerbations and a new GABHS infection. In the original PANDAS cohort, no individual had “overt chorea,” but all except one had choreiform movements and 50% had “marked choreiform” movements, leading some reviewers to suggest that the diagnosis was actually SC. This possibility has been enhanced by the finding of overlapping biomarkers for SC and children with PANDAS plus choreiform movements. In order to circumvent the issue of “associated with streptococci,” other authors have proposed terms such as CANS (children with acute neuropsychiatric symptoms) (67) or PANS (pediatric autoimmune neuropsychiatric symptoms) (69). As emphasized in the former above, a comprehensive investigation is essential for the proper diagnosis. A small treatment study for PANDAS showed IVIG therapy–improved OCD symptoms, but not tics, and plasma exchange improved both (70). Symptomatic therapy is recommended.
Acute Disseminated Encephalomyelitis
After a clinical pharyngitis with laboratory evidence of GABHS, 10 children developed behavioral changes, somnolence, stupor, and a movement disorder, including rigidity, dystonic posturing, or hemidystonia. No tics or chorea were reported. MRI demonstrated T2 hyperintense basal ganglia lesions in 8 of 10 patients, and there was a CSF pleocytosis in 7 of 10 patients. All 10 had elevated serum anti-basal ganglia antibodies (71).
Anti-NMDA Receptor Encephalitis
A recently emerging entity, anti-NMDA receptor encephalitis can present in multiple age groups, but presentations in children may be different from their adult counterparts. Movement disorders can be a hallmark of the disorder, with other presenting symptoms including psychiatric manifestations, seizures, and autonomic changes (72). Typical movements in children may include chorea, dystonia, myorhythmia, orolingual dyskinesia, or stereotypic movements, with some displaying multiple abnormal movements (73). The disorder has been associated with malignancy, specifically ovarian teratoma; however, younger patients are more likely to have a preceding infection than an associated tumor (72).
HEREDITARY AND METABOLIC DISORDERS ASSOCIATED WITH MOVEMENT DISORDERS
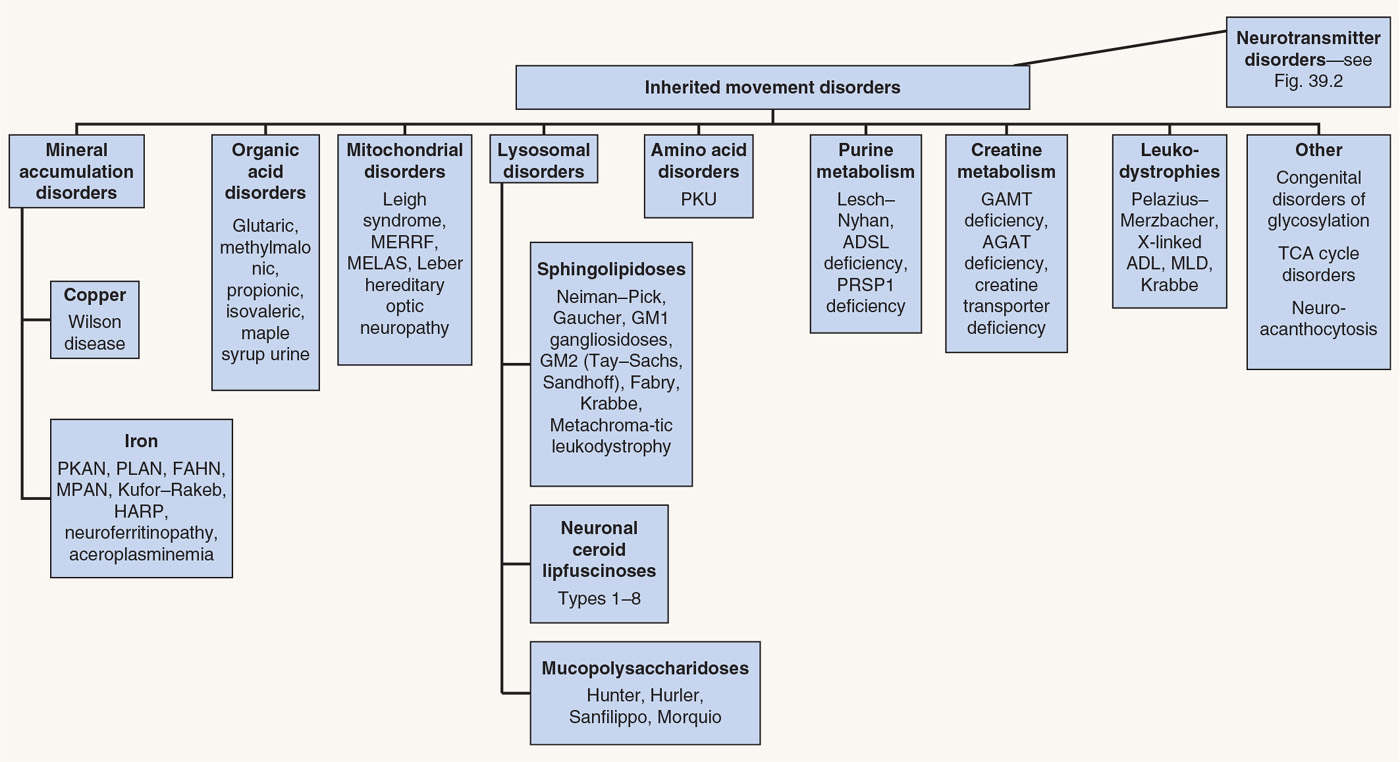
Movement abnormalities in children are frequently associated with hereditary metabolic disorders (Fig. 39.1). Inborn errors of metabolism are individually rare, but cumulatively cause significant morbidity and mortality.
PEDIATRIC NEUROTRANSMITTER DISEASES
The term “pediatric neurotransmitter disease” has been applied to relatively uncommon genetic disorders that affect the metabolic pathways of neurotransmitters. The primary neurotransmitters involved in these diseases include the monoamines serotonin, dopamine, and norepinephrine as well as GABA.
Monoamine-related neurotransmitter diseases can be divided into separate categories based on the site of abnormality in the metabolic pathway (Fig. 39.2) such as disorders affecting cofactors (tetrahydrobiopterin, BH4), enzymes of biosynthesis (tyrosine hydroxylase, TH, or aromatic amino acid decarboxylase, AADC), and those affecting enzymes of catabolism. Diagnostic studies include CSF analysis of monoamines and of neurotransmitter metabolites, including homovanillic acid (HVA), 5-hydroxyindoleacetic acid (5-HIAA), and 3-methoxy-4-hydroxylphenylglycol (MHPG). Quantitative measurement of catecholamines can also be obtained in urine and plasma (74).
< div class='tao-gold-member'>