INTRODUCTION
Movement disorders commonly occur in association with systemic diseases, and they may occasionally be the presenting feature of a systemic disease (1,2). It is therefore imperative to carefully consider patients’ known systemic diseases (if any), risk factors for the development of systemic disease, signs or symptoms suggestive of systemic disease, and all medical historical details, as these may provide important clues to the underlying etiology of abnormal movements. Identification and treatment of the underlying systemic disorder is essential for optimal management, though additional symptomatic therapy to suppress abnormal movements is often required. This chapter seeks to provide clinicians with a broad overview of the movement disorders occurring in association with several categories of systemic disease, including autoimmune, endocrine, metabolic, hematologic, toxic and nutritional, infectious, cerebrovascular, and paraneoplastic diseases. Some of these topics, such as paraneoplastic movement disorders, are the focus of other chapters within this book. The reader is strongly encouraged to refer to these other chapters as needed for more detail.
AUTOIMMUNE DISEASE
Autoimmune diseases arise from an overactive immune response against normal body tissue. They can be restricted to certain organs or tissue types, or can occur in a more generalized distribution. The autoimmune diseases most often associated with movement disorders are discussed below.
SYSTEMIC LUPUS ERYTHEMATOSUS
Possible neurologic manifestations of systemic lupus erythematosus (SLE) can include psychosis, seizures, encephalopathy, neuropathy, transverse myelitis, as well as a variety of movement disorders. Chorea, which occurs in 2% to 4% of cases, is the most commonly encountered movement disorder related to SLE; however, numerous others including cervical dystonia, blepharospasm, spasmodic torticollis, hemiballismus, tremor, parkinsonism, and stiff-person syndrome (SPS) have been reported (3,4). Chorea is usually accompanied by other neurologic manifestations of the disease, although it can sometimes be the presenting feature (5). Chorea is often asymmetric at presentation, but it typically evolves into a generalized distribution that can interfere with speech, balance, and activities of daily living. Typically, it spontaneously resolves after several days or weeks, though it can sometimes be permanent. Young women with SLE are most likely to develop chorea, and exacerbations have been reported in association with pregnancy and oral contraceptive use, suggesting an unclear role of estrogens in the development of chorea (3,6,7). Pathologically, widespread microinfarcts are the classic hallmark of neurolupus; however, this is inconsistently reported (8). Antiphospholipid antibodies, which predispose patients to recurrent thrombotic events, are strongly associated with chorea in SLE, though the mechanism remains unclear. Magnetic resonance imaging (MRI) of the brain is usually normal, although ischemic lesions in the basal ganglia may be found (8,9). The chorea may respond to low doses of haloperidol, tetrabenazine, valproic acid, or clonidine (3,10,11). Intravenous immunoglobulin (IVIg) and plasmapheresis have also reported to successfully treat refractory chorea in SLE (12).
ANTIPHOSPHOLIPID ANTIBODY SYNDROME
Primary antiphospholipid syndrome (APS) is defined by recurrent thrombosis and miscarriages in association with high titers of antibodies such as anticardiolipin IgG or IgM, lupus anticoagulant, or anti-B2-glycoprotein 1 (13). Chorea is the most commonly associated movement disorder and has been reported in 1.3% of patients (3,14). It clinically resembles SLE chorea. Imaging studies are often normal, supporting an immunologic rather than ischemic etiology. Successful treatment has been reported with the use of corticosteroids, anticoagulants, aspirin, and dopamine antagonist medications (3). Rare cases of hemidystonia, paroxysmal nonkinesiogenic dyskinesia, writer’s cramp, ataxia, corticobasal-like syndromes, and parkinsonism have also been reported in APS (3).
AUTOIMMUNE THYROID DISEASE
Antithyroperoxidase (anti-TPO) and antithyroglobulin (anti-TG) antibodies that alter thyroid function in autoimmune thyroid disease are also responsible for Hashimoto’s encephalopathy, which is also referred to as steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT). This condition results in an encephalopathy often accompanied by involuntary movements and ataxia. Interestingly, SREAT can occur despite a biochemically euthyroid state. The most commonly reported movement disorders associated with SREAT are chorea, tremor, multifocal myoclonus, and ataxia. However, opsoclonus, akinetic–rigid syndromes, palatal tremor, paroxysmal kinetic dyskinesia, and myorhythmia have also been reported (1,15). Brain MRI is usually normal or can show diffuse nonspecific white matter abnormalities (1,16). An abnormal electroencephalogram (EEG) has been reported in approximately 90% of cases, showing nonspecific changes and generalized slowing (16). Elevated CSF protein is common, and both mild pleocytosis and oligoclonal clonal bands can also occur (17). This condition is responsive to high-dose steroid treatment (1).
CELIAC DISEASE
Celiac disease (gluten enteropathy) is an autoimmune disease characterized by a dietary gluten sensitivity causing damage to the lining of the small intestine. Approximately 10% to 50% of patients with celiac disease experience neurologic symptoms (3). Gait ataxia is one of the most common neurologic manifestations (3,18,19), suspected to result from direct toxicity of antigliadin antibodies to the cerebellum, posterior columns of the spinal cord, and peripheral nerves (20,21). Other neurologic symptoms that have been reported include eye-movement abnormalities, dysarthria, limb ataxia, chorea, and rarely palatal tremor (3,22). Sensorimotor axonal neuropathy is also common (20). Brain MRI usually reveals cerebellar atrophy, both in the vermis and hemispheres out of proportion to any concurrent cerebral atrophy (21). Pathologically, perivascular cuffing is seen with both CD4 and CD8 cells in the white matter of the cerebellum along with significant Purkinje cell loss (23). Ataxia is typically not responsive to treatment. However, IVIg and gluten-restricted diets have been beneficial in some case reports (24). It is important to note that up to 85% of patients with biopsy-proven celiac disease do not report any gastrointestinal symptoms; thus, the absence of these symptoms cannot exclude the diagnosis especially in the presence of idiopathic progressive ataxia (21).
SJÖGREN’S SYNDROME
Sjögren’s syndrome is a chronic autoimmune disease characterized by destruction of the salivary and lacrimal glands. Between 1.5% and 25% of cases are associated with neurologic complications, including rare movement disorders (3). Parkinsonism is the most common movement disorder described in association with Sjögren’s syndrome. Clinically, patients have features of slowly progressive atypical parkinsonism with a symmetric akinetic–rigid syndrome, gait disorder, postural instability, and minimal tremor (25). Typically, these are not responsive to levodopa therapy (25). Other movement disorders occurring in Sjögren’s syndrome include cerebellar degeneration, chorea, and dystonia (26–28). Brain MRI can be normal or may reveal multiple symmetric T2 hyperintense lesions in periventricular, subcortical, and deep locations involving both grey and white matter (25,29). Pathophysiology remains unclear, and response to immunosuppressive therapy is inconsistent.
BEHÇET’S DISEASE
Behçet’s disease is an autoimmune disease characterized by a small vessel systemic vasculitis, often presenting with mucous membrane ulceration and ocular issues. It can cause a variety of neurologic problems, including rare movement disorders such as tremor, palatal myoclonus, chorea, oromandibular dystonia, and parkinsonism (30–33). CSF pleocytosis and elevated protein levels are common (33). MRI may be normal or may reveal focal lesions throughout the brain (33). Pathologically, there may be scattered foci of necrosis, demyelination, and scarring throughout the CNS (33). An inconsistent response to immunosuppressive therapy has been reported.
SARCOIDOSIS
Sarcoidosis is a systemic granulomatous disease that rarely presents with signs of central nervous system involvement (approximately 5% of cases), though up to 15% of autopsy cases have been reported to show evidence of neurosarcoidosis (34). Basilar meningitis is the most common neurologic presentation of neurosarcoidosis, though parenchymal lesions can also occur, producing a variety of neurologic symptoms (35). Granulomatous infiltration of the basal ganglia may cause parkinsonism, hemidystonia, chorea, or hemiballismus (35). The diagnosis of neurosarcoidosis can be challenging because no test, apart from biopsy, is completely reliable. Parenchymal lesions often show gadolinium enhancement on MRI. CSF protein is usually elevated, and pleocytosis and oligoclonal bands may be present (36). Additional tests such as CSF ACE levels are of little added value. Corticosteroids or immunosuppressants may improve abnormal movements, but results are inconsistent (36,37).
ENDOCRINE AND METABOLIC DISORDERS
THYROID DISEASE
Hyperthyroidism has been reported to induce or exacerbate a variety of hyperkinetic movement disorders. Enhanced physiologic tremor is the most common hyperkinetic disorder associated with hyperthyroidism, occurring in as many as 97% of hyperthyroid patients (38). Changes in peripheral β-adrenergic receptor tone may be the pathophysiologic basis, supported by the fact that β-blockers often effectively suppress the tremor. Other, less common movement disorders associated with hyperthyroidism are chorea, ballism, myoclonus, ataxia, and SPS (38,39). Brain MRI is usually normal (4). The movement disorders are reversible with treatment of the underlying hyperthyroidism, and often reemerge if hyperthyroidism redevelops (39). Hyperthyroid chorea may also respond to dopamine receptor–blocking agents which, in conjunction with the fact that there are decreased concentrations of the dopamine metabolite homovanillic acid in the CSF of hyperthyroid patients, may suggest that altered dopamine turnover or increased dopamine-receptor sensitivity could play a role in the pathophysiology (40).
Movement disorders are less often seen in association with hypothyroid states, the most common being truncal ataxia (41). Ataxia improves as the patient becomes euthyroid. There have also been a few case reports of chorea in hypothyroidism after levothyroxine replacement, resulting in “intracerebral hyperthyroidism” leading to the abnormal movements (42).
PARATHYROID DISEASE
Hypoparathyroid states can be associated with movement disorders such as paroxysmal dystonias, parkinsonism, or choreoathetosis (43–45). The pathophysiology is poorly understood. Associated hypocalcaemia can lead to tetany and neuromuscular irritability, inducing carpopedal spasm and Chvostek’s sign (46). Calcifications occur in the basal ganglia in most patients with primary hypoparathyroidism, but they do not correlate well with the presence of movement disorders (43,47). Improvement of the abnormal movements occurs with treatment of the underlying hypoparathyroidism and associated electrolyte abnormalities.
DIABETES MELLITUS
Choreoathetosis can rarely be caused by either hypoglycemia or hyperglycemia (48,49). When due to hypoglycemia, chorea usually resolves as blood sugar normalizes. Occasionally, however, recurrent hypoglycemic episodes have resulted in a permanent bilateral chorea (50). Acute nonketotic hyperosmolar hyperglycemia can result in a syndrome of hemiballism–hemichorea (HB–HC). Most reported cases have occurred in elderly women with blood glucoses in the 400- to 1,000-mg/dL range. HB–HC may disappear after resolution of the hyperglycemia, or it may persist for months or even indefinitely (50–54). Brain MRI often reveals T1 contralateral putaminal hyperintensity (51). The pathophysiology of this disorder is uncertain, although dopamine hypersensitivity in postmenopausal women, coexistence of lacunar infarction in the contralateral basal ganglia, and increased blood flow in the contralateral striatum and thalamus have all been suggested as possible pathogenic factors (53,54). Treatment with dopamine-receptor antagonists or tetrabenazine can be effective if symptoms do not quickly resolve with normalization of blood glucose levels (51,55). Fig. 42.1 features a typical case.
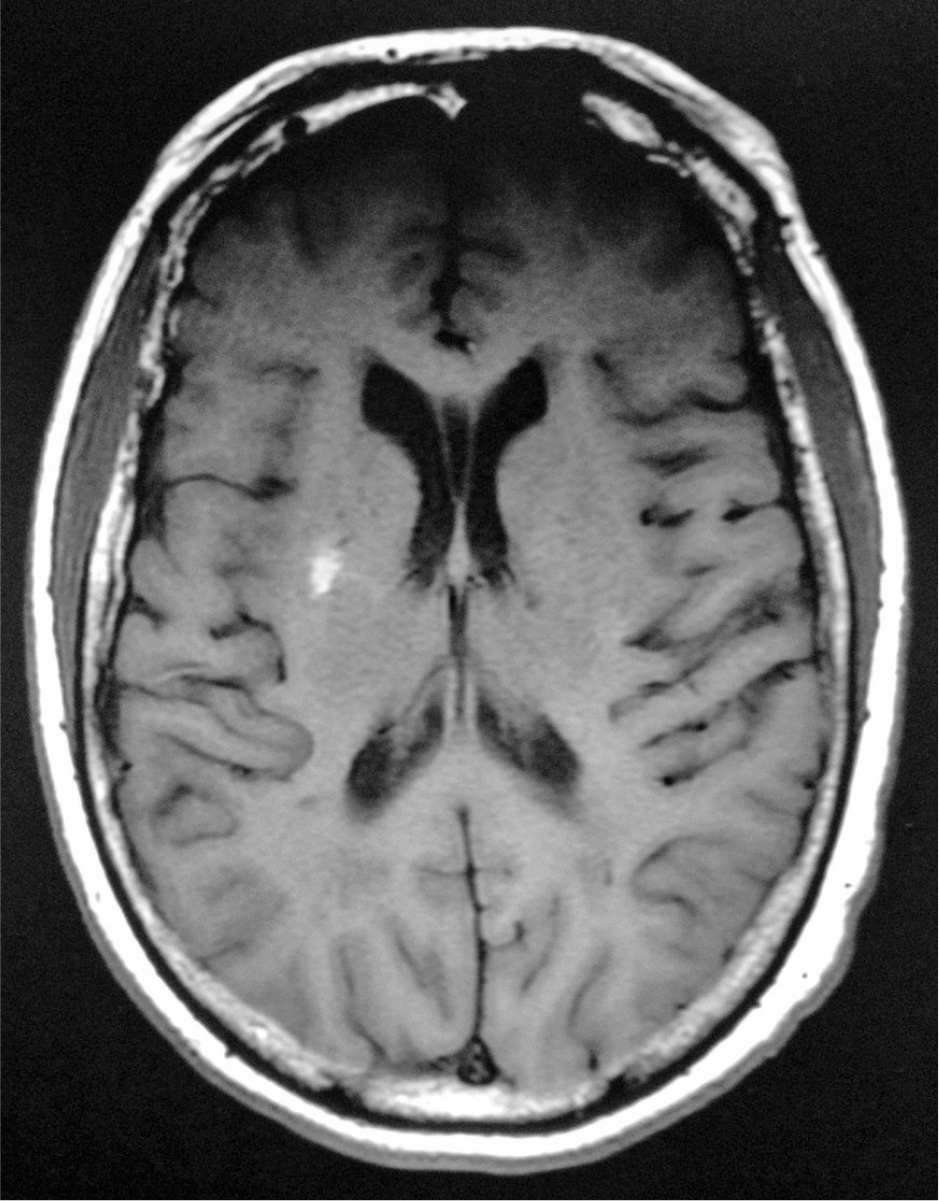
Figure 42.1. MRI from a 62-year-old type 1 diabetic woman presenting to the emergency room with hemichorea–hemiballismus (Image courtesy of Dr. Binod Wagle). Note the T1 hyperintense lesion in the right putamen. Her chief complaint was that she could not stop her left arm from moving. She had experienced sudden onset of the involuntary movements 2 weeks prior to presentation. The movements were intermittent initially, but they soon became continuous. Exam revealed choreiform and ballistic movements of the left upper extremity. She had been intermittently noncompliant with her diabetic medication regimen for many months, and her blood glucose levels had been consistently running in the 400s at home during the weeks prior to presentation. Blood glucose upon arrival was 847 mg/dL. Hemoglobin A1C was 12.8. The movements resolved during her hospitalization with normalization of her blood glucose levels.
RENAL DISEASE
Multiple movement disorders such as tremor, ataxia, asterixis, myoclonus, and restless legs syndrome (RLS) can occur in association with renal failure and uremic encephalopathy. A coarse, irregular postural and action tremor is often seen, which may respond partially to β-blockers (56). Asterixis and multifocal myoclonus (either action or stimulus-sensitive) are also commonly reported in uremic encephalopathy (56–59). Benzodiazepines are often an effective treatment for myoclonus. RLS occurs in 15% to 20% of patients with chronic kidney disease (60), usually in association with iron deficiency. Dopamine agonists and levodopa are used as first-line treatments for RLS, followed by second-line agents such as benzodiazepines, gabapentin, clonidine, and opioids. Correction of underlying iron deficiency is also helpful. Lastly, RLS has been reported to improve dramatically after kidney transplantation (61,62).
LIVER DISEASE
Hepatic encephalopathy is the most common neurologic complication of advanced liver disease. While movement disorders such as tremor, asterixis, and myoclonus do occur in hepatic encephalopathy, they are relatively uncommon (63). Movement disorders are more often observed in patients with acquired hepatocerebral degeneration (AHD), a chronic and progressive syndrome characterized by parkinsonism, ataxia, dystonia, chorea, or orobuccolingual stereotypy (similar to tardive dyskinesia) (64). The onset of AHD is subacute and the course highly variable, often not paralleling other features of advancing liver disease. In contrast with hepatic encephalopathy, the abnormal movements occurring with AHD are not associated with a depressed level of consciousness and do not respond to ammonia-lowering agents (63). AHD can occur in patients with advanced liver disease from any cause, but is reported more frequently in those with evidence of portosystemic shunting. Rare cases have been reported in patients with portosystemic shunting in the absence of underlying hepatocellular disease, such as those with portal vein thrombosis (65). It is suspected that portosystemic shunting may lead to AHD by allowing neurotoxins to bypass hepatic metabolism and enter the brain via the systemic circulation (66). While the precise neurotoxin is unknown, ammonia, aromatic amino acids, and manganese have been suggested as candidates (66). The prevalence of AHD is unknown, though movement disorders are estimated to occur in 20% to 90% of patients needing liver transplantation (64). In contrast with idiopathic Parkinson’s disease, the parkinsonism associated with AHD tends to be symmetric, rapidly progressive, and is more likely to be associated with cognitive impairment and early postural instability (67). Action tremor is more common than rest tremor. Pathologically, patchy degeneration of neurons with microcavitation and pseudolaminar necrosis in the deep cortical layers and basal ganglia have been described (66,68). Brain MRI often shows increased T1 signal in the pallidal nuclei. T2 and postcontrast images are usually normal (63,64). Hyperkinetic movements such as chorea and orobuccolingual stereotypies can be treated with dopamine-receptor antagonist medications or tetrabenazine (11). Levodopa and dopamine agonists may be useful in some patients with parkinsonism, though the response is variable (64,69). AHD and the associated movement disorders often improve following liver transplantation (70). Fig. 42.2 features a case of AHD with typical MRI findings.
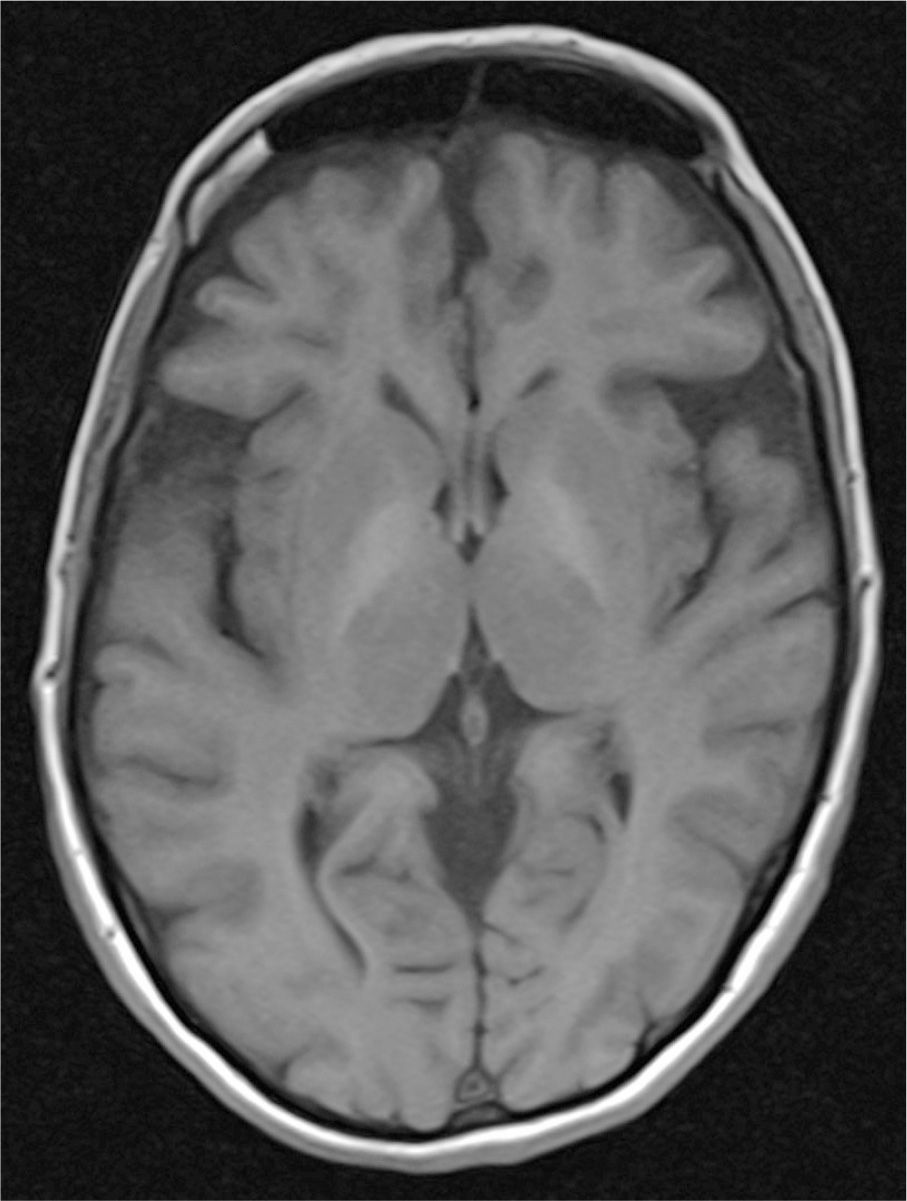
Figure 42.2. MRI from a 62-year-old woman with acquired hepatocerebral degeneration. Note the T1 hyperintense lesions bilaterally in the basal ganglia. She presented to the movement disorders clinic complaining of hand tremors for 6 months. She had a history of protein C deficiency, portal vein thrombosis, and chronic pancreatitis. At the time of her initial evaluation, her liver enzymes were normal, as was her ammonia. Examination revealed mild, symmetric parkinsonism with a postural and kinetic tremor of her bilateral upper extremities. She also had mild blepharospasm.
HEMATOLOGIC CONDITIONS
Polycythemia vera (PCV) is a myeloproliferative disorder in which there is overproduction of red blood cells from the bone marrow. The most commonly reported movement disorder seen with PCV is chorea; however, it is rare and only reported in less than 1% of patients. The chorea is more often seen in females and is typically bilateral and symmetric (71). It is suspected that the chorea results from blood hyperviscosity, causing small infarctions or hemorrhage in the basal ganglia (71). The chorea may improve with treatment of the underlying PCV; however, reserpine or tetrabenazine are also often used (71). While there are some isolated case reports of movement disorders occurring in association with other hematologic disorders such as porphyria, there is insufficient information in the medical literature to enable further discussion of these possible associations (72).
TOXIC AND NUTRITIONAL DEFICIENCIES
VITAMIN E MALABSORPTION
Tocopherol (vitamin E) deficiency can be seen in patients with any systemic disease causing fat malabsorption (e.g., cystic fibrosis). Chronic vitamin E deficiency may lead to a syndrome of spinocerebellar ataxia and peripheral neuropathy with associated symptoms of dysarthria, oculomotor abnormalities, and loss of vibratory and position sense. Pathologically, loss of myelinated nerve fibers in the posterior columns and CNS occurs (73). Oral or parenteral vitamin E supplementation should be initiated to prevent progressive neurologic decline.
ETHANOL
Although alcohol may transiently alleviate certain movement disorders such as essential tremor and myoclonus–dystonia syndrome, it can also trigger or exacerbate others, or even cause a variety of movement disorders associated with its withdrawal (74). Alcohol withdrawal in physically dependent individuals is associated with a distal, coarse, irregular tremor worse with movement that is quickly improved with alcohol ingestion or benzodiazepine use. If left untreated, delirium tremens may develop (75). Transient parkinsonism can develop in the setting of alcohol withdrawal. It often remits spontaneously in a few days or weeks (76). Transient orolingual dyskinesias, occasionally also involving neck and arm muscles, have also been reported in the setting of alcohol withdrawal (77).
A high frequency postural tremor can be found in alcoholics who are not actively drinking or withdrawing. β-Blocking drugs may be effective in suppressing this tremor. A 3-Hz tremor of the legs that is slow and rhythmic and involves flexion and extension of the hip girdle muscles can also be seen in alcoholics and can affect gait (77). Chronic alcohol abuse can lead to alcoholic liver disease, which can ultimately cause acquired hepatic encephalopathy or AHD, both of which are associated with movement disorders discussed earlier in this chapter. Truncal ataxia can be seen with acute alcohol intoxication as well as in chronic alcohol dependence. Pathologically, degeneration of the anterior superior aspect of the vermis and possibly atrophy of the cerebellar hemispheres in severe cases are present (78). Significant loss of Purkinje cells is typical (78).
INFECTIOUS DISEASES
VIRAL INFECTIONS
Prospective studies suggest that 5% to 44% of HIV/AIDS patients may suffer from movement disorders, including hemichorea–hemiballismus, myoclonus, dystonia, tremor, parkinsonism, and akathisia (79). The pathogenesis of these movement disorders is highly variable and can result directly from viral invasion of the basal ganglia, from acquired concurrent opportunistic infections, or from other complications of the disease such as primary CNS lymphoma, AIDS–dementia complex (ADC), or progressive multifocal leukoencephalopathy. HIV patients are also significantly more susceptible to the development of extrapyramidal reactions when exposed to neuroleptic medications (80). Table 42.1 summarizes the movement disorders occurring in association with HIV/AIDS.
As previously mentioned, HIV can directly invade the basal ganglia. Immunocytochemistry studies have revealed high viral loads in the basal ganglia and substantia nigra, and SPECT and PET studies have demonstrated abnormal metabolic activity of the basal ganglia in HIV patients (81,82). Pathologically, severe neuronal loss, reactive gliosis, and macrophage infiltration in the basal ganglia is seen (82).
| HIV/AIDS-Associated Movement Disorders |
Movement Disorder | Comments | Etiologies |
Dystonia | Rare | ADC CNS toxoplasmosis Neuroleptic-induced |
Hemichorea–ballism | Most common movement disorder in HIV/AIDS | ADC HIV encephalitis Opportunistic infections (CNS toxoplasmosis most commonly, Cryptococcus, PML) |
Myoclonus | Spinal, segmental, or generalized | ADC HIV infection Opportunistic infections (CNS toxoplasmosis, tuberculosis, herpes zoster, PML) |
Parkinsonism | May be atypical, symmetric, lacking rest tremor, with early postural instability | ADC HIV infection Neuroleptic-induced Opportunistic infections (CNS toxoplasmosis, tuberculosis, Whipple’s disease, PML) |
Paroxysmal dyskinesias | Kinesiogenic or nonkinesiogenic | ADC |
Tics | Rare | CNS toxoplasmosis |
Tremor | Most often a mild, bilateral postural tremor | ADC HIV infection Neuroleptic-induced Opportunistic infections (CNS tuberculosis) TMP-SMX induced |
HIV, human immunodeficiency virus; AIDS, acquired immunodeficiency syndrome; ADC, AIDS dementia complex; CNS, central nervous system; PML, progressive multifocal leukoencephalopathy; TMP-SMX, trimethoprim–sulfamethoxazole. Source: Adapted from Tse W, et al. Movement disorders and AIDS: a review. Parkinsonism Relat Disord 2004;10:323–334. | ||
Aside from HIV/AIDS, many other viral infections can be associated with the development of movement disorders, though a detailed discussion of each one is beyond the scope of this chapter. Table 42.2 lists the viruses known to cause movement disorders.
BACTERIAL INFECTIONS
Bacterial infections can lead to movement disorders through both antibody-mediated and direct mechanisms. Examples of both mechanisms are reviewed below.
ANTIBODY-MEDIATED DISORDERS
Infection with Group A β-hemolytic Streptococcus (GABHS) can be associated with the development of Sydenham’s disease (SD). SD usually appears 1 to 6 months after the acute pharyngitis. Due to the widespread use of antibiotics, SD is now rare in developed countries. SD is most common between the ages of 8 and 9 years old, and it is more common in females (83). SD is typically characterized by generalized chorea, though hemichorea has been described in 20% of cases (83). Other features can include ballism, facial grimacing, tics, dysarthria, oculomotor abnormalities, motor impersistence, gait abnormalities, seizures, hyperactivity, obsessions, compulsions, anxiety, and even psychosis (83).
High titers of antistreptolysin are typical. MRI is usually normal, though enlargement of the caudate, putamen, and globus pallidus are sometimes seen (4). Reversible striatal hypermetabolism may be seen on PET and SPECT studies (83). Pathologically, there is widespread cell loss in the basal ganglia, frontal and temporal cortices, and small vessel vasculitis (83). Accurate diagnosis and proper antibiotic treatment of acute pharyngitis is essential to prevent SD. Symptomatic treatment with valproic acid or neuroleptic medication is used until the condition spontaneously resolves. Prednisone has also been shown to be effective in a double-blind placebo-controlled study (84).
Streptococcal infections can also lead to the development of the PANDAS (pediatric autoimmune neuropsychiatric disorders after streptococcal infections) syndrome, involving the presence of obsessive–compulsive or tic disorder with prepubertal onset, abrupt onset with significant exacerbations, temporal association with GABHS infection, and associated neurologic abnormalities. It is unclear and controversial whether PANDAS is simply a variant of SD or a separate entity (85). The reader is encouraged to refer to Chapter 21 of this text, which discusses SD in greater detail.
DIRECT MECHANISMS
CNS tuberculosis can cause a variety of movement disorders such as tremor, chorea, ballism, dystonia, myoclonus, and parkinsonism. Both tuberculous meningitis and intracranial tuberculomas can cause movement disorders. In fact, movement disorders may occur in up to 16.6% of tuberculous meningitis cases and 30% of patients with intracranial tuberculomas (1,86). In tuberculous meningitis, neuroimaging may be normal or can show ischemic or hemorrhagic infarcts in the basal ganglia or diffuse lesions at the diencephalic–mesencephalic level (1). Hydrocephalus is commonly present (1). While chorea and dystonia are associated with deep tuberculomas, tremor is more often associated with surface lesions (86). Tremor and myoclonus can occur in spinal tuberculosis as well (1). Diagnosis is usually made from culture of Mycobacterium tuberculosis from CSF, although CSF PCR may also be used. Therapy includes antituberculosis drugs for a minimum of 6 months. Movement disorders generally improve within a few weeks.
| Viral Causes of Movement Disorders |
Movement Disorder | Viruses |
Chorea | Cytomegalovirus Encephalitis lethargica Enterovirus Epstein–Barr Herpes simplex Influenza Japanese encephalitis Measles Paramyxovirus Rubella Varicella-zoster |
Dystonia | Influenza Japanese encephalitis |
Myoclonus | Encephalitis lethargica Measles (subacute sclerosing panencephalitis) |
Oculogyric crisis | Encephalitis lethargica |
Opsoclonus–myoclonus | Varicella-zoster |
Parkinsonism | Arbovirus Encephalitis lethargica Enterovirus Influenza Japanese encephalitis Measles Varicella-zoster |
Tics | Herpes simplex |
Tremor | Influenza Japanese encephalitis |
Source: Adapted from Cardoso, F. Infectious and transmissible movement disorders. In: Jankovic J, Tolosa E, eds. Parkinson’s Disease and Movement Disorders. 3rd ed. Baltimore: Williams & Wilkins, 1998:945–966. | |
Whipple’s disease (WD) is a rare multisystem illness caused by Tropheryma whipplei infection. Symptoms include malabsorption with steatorrhea, arthralgia and arthritis, lymphadenopathy, and rarely neurologic symptoms (including dementia, visual impairment, papilledema, supranuclear ophthalmoplegia, seizures, myoclonus, ataxia, and a depressed level of consciousness). Oculomasticatory myorhythmia, which is unique to WD, is characterized by slow, rhythmic, synchronous, repetitive contractions that occur in ocular, facial, and masticatory muscles (87). Ocular myorhythmia presents with continuous, horizontal, pendular, small-amplitude, vergence oscillations of the eyes, occurring approximately every second. Movements of the face and jaw may be quicker and resemble rhythmic myoclonus. These movements may persist in sleep. While jejunal biopsy was required for diagnosis in the past, PCR techniques are now typically used for diagnosis. Jejunal biopsy shows macrophages laden with periodic acid–Schiff–positive granules and bacilli (88). Current recommended treatment is parenteral administration of streptomycin and penicillin G or ceftriaxone for 2 weeks followed by oral administration of trimethoprim–sulfamethoxazole for 1 to 2 years (88).
Movement disorders may also occur in the course of bacterial meningitis caused by a variety of organisms, including Mycoplasma pneumoniae, Salmonella, Legionella pneumophila, Borrelia burgdorferi, Treponema pallidum, Haemophilus influenzae, Streptococcus pneumonia, and Neisseria meningitides (4). The pathogenesis of these movement disorders is poorly understood, and a detailed discussion of each causative agent is beyond the scope of this chapter.
PARASITIC INFECTIONS
Falciparum malaria infections can be complicated by movement disorders. Acute or delayed cerebellar syndromes with opsoclonus and tremor can occur (89). Opsoclonus–myoclonus syndrome, chorea, hemiballismus, dystonia, and parkinsonism can also occur (4,90). The pathophysiologic mechanisms are not well understood. MRI images are usually normal. Antimalarial treatments are essential. Opsoclonus–myoclonus syndrome has been reported to respond well to oral clonazepam (90).
Neurocysticercosis (NCC) is caused by the encysted larvae of the Taenia solium tapeworm. Movement disorders, most commonly parkinsonism and tremor, occur in 3.5% of cases of NCC (1). Chorea, myoclonus, hemifacial spasm, and dystonia have also been described (1). Cysts in the basal ganglia and related structures are the presumed pathogenic mechanism. Chorea and dystonia are associated with deep lesions, while tremor is associated with surface lesions (1). Parkinsonism can be associated with hydrocephalus (1). Movement disorders improve after treatment with albendazole, praziquantel, or surgery (1).
| Localization and Treatment of Poststroke Movement Disorders |
| Most Common Localization | Treatment |
Akathisia | Posterior thalamus | Clonazepam |
Asterixis | Thalamus, basal ganglia, frontoparietal cortex, cerebellum, brain stem | Spontaneous resolution within a few days is typical |
Cerebellar outflow tremor | Posterior thalamus, or lesions of the dentatorubrothalamic pathway | Propranolol, primidone, clonazepam, valproic acid, levetiracetam, anticholinergics, levodopa (if resting tremor), adding weight to affected limb, DBS of Vim or Vop |
Chorea | Lentiform nucleus or thalamus | Neuroleptics, tetrabenazine, clonazepam, valproic acid, topiramate, stereotactic lesion or DBS of Vop or Vim |
Dystonia | Lenticular, especially putamen | Botulinum toxin, anticholinergics, benzodiazepines, baclofen, neuroleptics, tetrabenazine, stereotactic lesion, or DBS of globus pallidus or thalamus |
Holmes tremor | Dentatorubrothalamic, cerebellothalamic, or nigrostriatal pathways | Same as for cerebellar outflow tremor |
Hemiballism | Thalamus or striatum more often than subthalamus | Same as for chorea |
Myoclonus | Midbrain, pons, or thalamus | Clonazepam, valproic acid, levetiracetam, primidone, tetrabenazine, acetazolamide, botulinum toxin |
Stereotypies | Parietal, lenticulo–striatal, thalamic, midbrain, or left middle cerebral artery territory | Clonazepam, tetrabenazine |
Tics | Striatum, globus pallidus, frontal or parietal cortex | Clonidine, neuroleptics |
Transient dyskinesias | Severe carotid or vertebrobasilar stenosis | Endarterectomy |
DBS, deep brain stimulation; Vop, nucleus ventralis oralis posterior of the thalamus; Vim, nucleus ventralis intermedius of the thalamus. Source: Adapted from Mehanna R, Jankovic J. Movement disorders in cerebrovascular disease. Lancet Neurol 2013;12:597–608. | ||
CEREBROVASCULAR DISEASE
Ischemic or hemorrhagic strokes can result in a variety of movement disorders. The frequency of poststroke movement disorders is estimated to be 1% to 4% (91). Men and women are equally affected (91,92). Small vessel disease is the most common stroke mechanism associated with movement disorders, though cardioembolism, watershed events, as well as large and medium vessel atherosclerosis can also lead to movement disorders. Intracerebral hemorrhages are less frequently implicated (91).
The spectrum of movement disorders reported after cerebrovascular events is broad and includes parkinsonism, chorea–ballism–athetosis, dystonia, tremor, myoclonus, asterixis, transient limb shaking, stereotypies, akathisia, and tics (91). Abnormal movements can occur immediately at stroke onset, or they may emerge later. Dystonia is the most frequent delayed movement disorder reported after stroke, while chorea and hemiballism are most frequently seen in the acute stages of stroke (92). Conditions that predispose individuals to recurrent cerebrovascular events are worthy of consideration. For example, chorea and hemiballism have been reported in Moyamoya disease, a type of intracranial vasculopathy (93). Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) can lead to vascular parkinsonism (VP), which is discussed below (94). Table 42.3 features the most common anatomic localizations for poststroke hyperkinetic movement disorders, as well as their treatment options.
VP classically presents with predominantly lower-body involvement, manifested as a broad-based, shuffling, and often freezing gait with postural instability. Absence of a rest tremor and bilateral involvement are also typical (91). Commonly associated features include pyramidal tract signs, pseudobulbar palsy, and dementia (92). When compared to idiopathic Parkinson’s disease, VP often affects patients of older age with multiple vascular risk factors, tends to have a more rapid progression, and does not consistently respond well to levodopa (92). Lesions most commonly reported on MRI imaging are multiple lacunar infarcts affecting the basal ganglia or the supplementary motor area supplied by the anterior cerebral artery (92). Levodopa improves symptoms in one-third to half of cases (91). Fig. 42.3 features a typical case.
PARANEOPLASTIC SYNDROMES
Paraneoplastic neurologic syndromes are a heterogeneous group of disorders. These syndromes are not caused by the tumor itself or metastatic disease, but rather from paraneoplastic antibodies simultaneously targeting antigens expressed by tumor cells as well as cellular components of the central and/or peripheral nervous system(s). The underlying cancer is often occult and the paraneoplastic neurologic syndrome may be the presenting complaint (2). Fortunately, paraneoplastic neurologic syndromes are rare, occurring in less than 1% of all cancer patients (95). The classical paraneoplastic neurologic syndromes that have been associated with the development of movement disorders are cerebellar degeneration (paraneoplastic ataxia), SPS, and opsoclonus–myoclonus syndrome. Much less commonly, paraneoplastic chorea, tremors, and parkinsonism can occur (95). Antibodies to the N-methyl-D-aspartate (NMDA) subtype of the glutamate receptor have been associated with a syndrome consisting of neuropsychiatric symptoms, dystonia, and other dyskinesias and is typically found in young women having an ovarian teratoma. A more extensive discussion of these movement disorders–caused paraneoplastic syndromes can be found in Chapter 33.
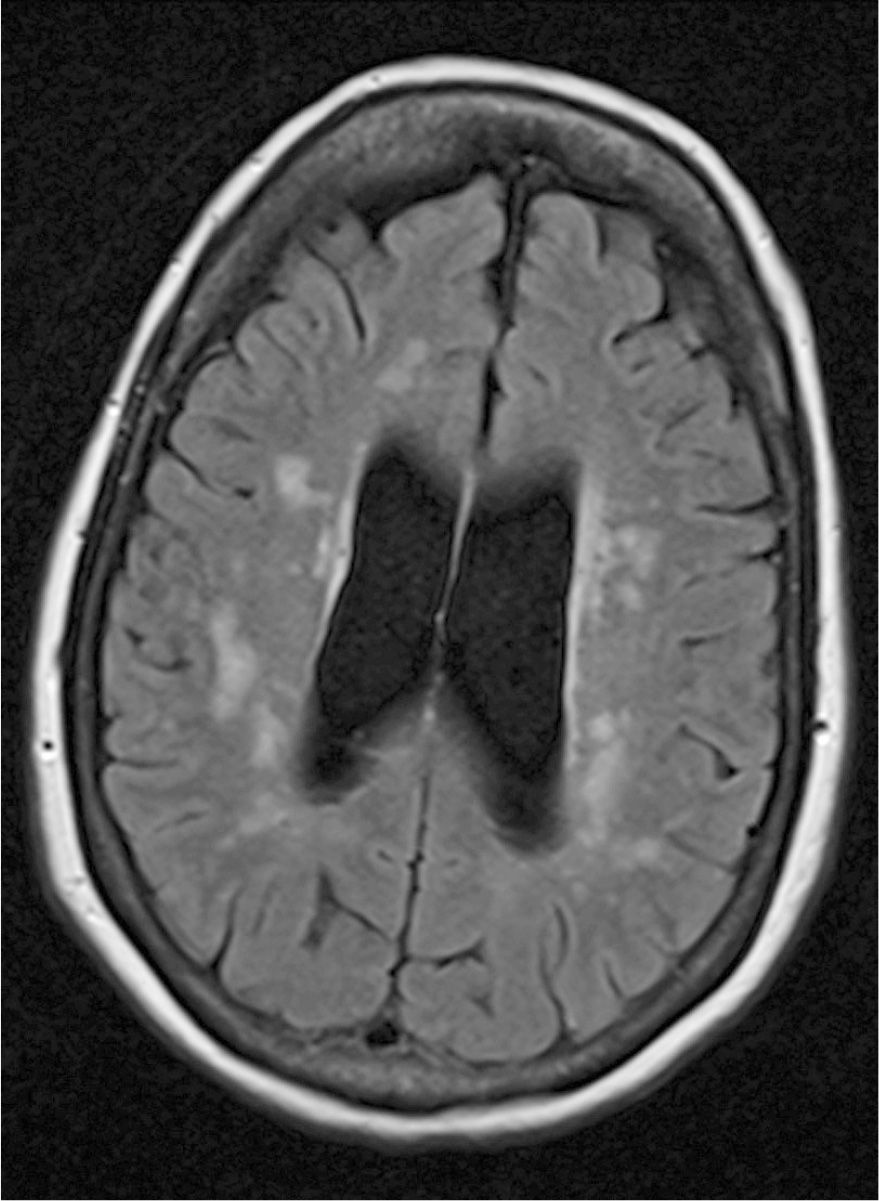
Figure 42.3. MRI from a 65-year-old woman with vascular parkinsonism (Image courtesy of Dr. Mark Baron). Note the extensive subcortical and periventricular white matter disease. She presented to the movement disorders clinic complaining of gait dysfunction with occasional falls. More specifically, her gait had slowed down, and she was experiencing freezing with turns. She endorsed feeling as though “my legs will not respond the way I want.” Onset was insidious over 1 to 2 years. There was a concomitant decline in her short-term memory. Past medical history was significant for coronary artery disease and ischemic optic neuropathy. Exam revealed prominent masked facies, bilateral brady/hypokinesia with finger taps, mild difficulty rising from seated position, and a moderately slow gait with shortened strides and poor arm swing. Reflexes were brisk throughout. A levodopa trial at a maximum dose of 600 mg daily was eventually aborted because it did not improve her symptoms and was causing significant gastrointestinal side effects.




