Multiple sclerosis (MS) is an immune-mediated, multiphasic, mutifocal disease of the central nervous system.
The primary etiology of MS is unknown; however the disease has features of inflammation, demyelination, axonal injury, and neurodegeneration.
Visual dysfunction, sensory disturbances, and gait impairment are very common presenting symptoms.
MRJ is the single most useful test in confirming the diagnosis of MS.
Early and accurate diagnosis is paramount.
Initiating early disease-modifying treatment can influence the clinical course.
Symptomatic therapy can greatly improve MS patients’ quality of life.
Multiple sclerosis (MS) is a neurodegenerative disease with early intense inflammation of the central nervous system (CNS). The primary etiology of MS remains unknown and is likely multifactorial. The disease is characterized pathologically by inflammatory infiltrates and demyelination, followed by varying degrees of secondary axonal degeneration.
▪SPECIAL CLINICAL POINT: Multiple sclerosis is the most common nontraumatic cause of neurologic disability in young adults and affects approximately 400,000 people in the United States.An estimated 200 people per week are newly diagnosed.
MS affects women two to three times more commonly than men. Most patients start with a period of unpredictable relapses and remissions, which in the majority is followed by an accumulation of neurologic dysfunction and a chronic progressive course. The life expectancy has been shown to be only 6 to 7 years less than that for a control population without MS, but the emotional and economic cost to society as a result of the disability is enormous.
HISTORY
The earliest account of a disease that appears likely to have been MS is found in writings from the 14th century, describing the illness of a Dutch nun, “Blessed Lidwina of Schiedam.” The earliest pathologic descriptions by Carswell and Cruveilhier date to between 1838 and 1845. Charcot is generally credited with the first comprehensive account of the clinical and pathologic features of MS, which was published in 1868.
EPIDEMIOLOGY
Epidemiologic studies have shown that MS has an unequal geographic distribution, with large regional and ethnic variations in the prevalence of disease (Fig. 11.1). MS is rare in the tropics and increases in frequency at higher latitudes north and south of the equator. The prevalence in the United States is reported at 57.8 per 100,000 and is almost twice as common in the Northern as compared to the Southern United States. The prevalence rate has been increasing, probably because of better recognition of MS and improved treatment of complications with a correspondingly increased longevity of those affected. Prevalence rates of less than 5 per 100,000 are found in Asia, Africa (except English-speaking whites in South Africa), and northern South America. MS is also said to be much less common among Eskimos, Gypsies, and African Americans. However, these patterns may be changing with increased travel; cases of MS have been reported in native Africans who have not traveled out of the country, and the prevalence of African Americans with MS has not been reexamined adequately in the magnetic resonance imaging (MRI) era of diagnosis.
FIGURE 11.1 Map of the world depicting areas of high prevalence (30+/100,000, solid), medium prevalence (5-29/100,000, dotted), and low prevalence (0-4/100,000, dashed). White areas are regions without data or people. (From Kurtzke JF, Wallin MT Epidemiology In: Burks JS, Johnson KR eds. Multiple sclerosis: diagnosis, medical management, and rehabilitation. New York: Demos Medical Publishing; 2000. Reproduced with permission from Demos Medical Publishing.)
Migration studies suggest that the risk of acquiring MS is related to the location in which one has lived before puberty. Individuals migrating from high- to low-risk areas decreased their expected risk of developing MS, as determined from their area of birth. Conversely, migration from low- to high-risk areas increased the risk of acquiring MS. The reliability of migration studies has been questioned, and no definite conclusions can be made from these data.
Numerous instances of MS clusters have been reported. Several clusters have been related to exposure to heavy metals and canine distemper virus, but no conclusive link has been established. Genetic studies strongly suggest that the disease is polygenic and that genetic factors may have a stronger influence in determining susceptibility to MS than environmental factors.
PATHOLOGY OF MULTIPLE SCLEROSIS
Classic descriptions of the MS lesion as predominantly demyelinating with relative sparing of axons still hold true today. Modern tools have afforded more detailed descriptions of the immunologic and structural events occurring within lesions and have reemphasized the secondary axonal degenerative stage of the lesion. Major advances in the classification of MS pathology have been made from biopsy and rapid autopsy material. Clinicopathologic correlations using MRI and spectroscopy provide hope that advances in our understanding of the pathologic state will allow guided therapeutic interventions.
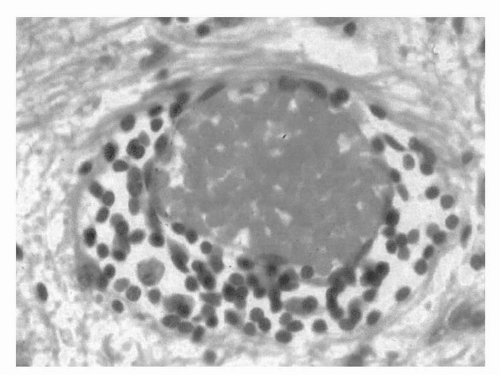
FIGURE 11.2 Perivenular infiltrate typical of the early acute inflammatory event in an active lesion.
FIGURE 11.3 Luxol fast blue stain revealing loss of myelin in several irregular geographic lesions.
The MS plaque appears to begin with the migration of lymphocytes and macrophages across the blood-brain barrier into the perivenular space (Fig. 11.2). This is followed by diffuse parenchymal infiltration by inflammatory cells, edema, and active stripping of myelin from axons by macrophages, leading to multifocal areas of demyelination (Fig. 11.3). Subsequently, astrocytic hyperplasia and the accumulation of lipid-laden macrophages ensue. As plaques enlarge and coalesce, the initial perivenular distribution of the lesions becomes less apparent. The inflammatory reaction is usually less pronounced in grey matter, probably because of the smaller amount of myelin in these areas. The extent of axonal loss in the demyelinated areas is highly variable. The sparing of axons is relative, and some axonal loss occurs in almost all lesions and can become substantial in severe cases. Axonal loss can also occur very early and even be present during the first clinical attack.
Plaquelike areas of pale myelin staining are called shadow plaques and are generally regarded as evidence of partial remyelination. Several studies have confirmed a remarkable potential for oligodendrocyte proliferation and partial remyelination, which seems to be impeded by as-yet-unknown local factors.
Myelin and Nerve Conduction
Proteolipid protein (PLP), myelin basic protein (MBP), and their isoforms make up 80% to 90% of the myelin sheath with 2′,3′-cyclic nucleotide 3′-phosphohydrolase (CNPase), myelin-associated glycoprotein (MAG), myelin oligodendrocyte protein (MOG), and other minor proteins making up the rest. Both PLP and CNPase are restricted to CNS myelin, whereas MBP constitutes about 10% of the protein in PNS myelin. Lipids constitute 80% of the dry weight of myelin. Cultured oligodendroglial cells contain a family of gangliosides of which GM3 and GM1 are the principal components.
One oligodendrocyte usually forms intern-odal segments of myelin on several different nerve fibers. This differs from the peripheral nervous system, where one Schwann cell contributes myelin to only one internodal segment. This difference may account, in part, for the much greater efficiency of regeneration in peripheral myelin. The nodes of Ranvier have a high concentration of sodium channels concentrated in the nodal membrane, which are required for saltatory conduction. During an episode of inflammatory demyelination, conduction may be transiently impaired as a result of edema, loss of myelin, and some degree of metabolic dysfunction of the nerve axon. Conduction can recover acutely on resolution of edema, subacutely with redistribution of sodium channels along the internodal membrane, or chronically after partial remyelination.
Pathologic Correlations with Magnetic Resonance Imaging
▪SPECIAL CLINICAL POINT: The profound impact of modern imaging technologies, particularly MRI.is evident from its expanding role in the diagnosis and prognosis of MS.
High signal (bright) T2-weighted lesions correlate well with the presence of lesions on gross pathology. However, T2 lesions lack specificity for microscopic tissue pathology and seem to represent a composite of factors including edema, demyelination, remyelination, gliosis, and axonal loss. Therefore, the T2-lesion burden does not correlate strongly with clinical disability even in clinically eloquent areas of the CNS (brainstem and spinal cord) because not all T2 lesions purport the destructive axonal pathology. Indeed, this is exactly what the T2-weighted lesion volume studies show—a weak but significant correlation with clinical disability (Expanded Disability Status Scale, or EDSS, and cognitive impairment). Because of the imprecise specificity of T2 images, more emphasis is being placed on Tl-weighted images in which persistent low signal (black holes) seems to correlate better with axonal dropout and clinical disability. However, one must be cautious because during the acute enhancing phase of a lesion, and perhaps for several months thereafter, low signal Tl lesions may represent extracellular edema that can resolve.
Another misunderstood aspect of imaging is that there is a temporal sequence of lesion pathology such that acute contrast-enhanced lesions correlate well with the perivenular infiltrate and the likelihood of a clinical exacerbation but also predict future formation of black holes and brain atrophy. Indeed in a clinical trial of interferon beta (IFNP), the drug suppressed inflammation on the MRI in the first year of treatment but did not slow brain atrophy until the second year. This suggests that those axons that were already demyelinated prior to treatment may follow an inexorable course of degeneration during the first year of treatment, but the axons that were salvaged from demyeli-nation by suppression of the inflammatory attack in the first year are spared degeneration in the second year. This also would explain why in two recent short-term trials of potent immunosuppressive drugs used for 12 months or one dose, contrast-enhancing lesions were reduced, but there was no effect on progression of disability or brain atrophy. Newer methods such as magnetization transfer imaging, diffusion tensor imaging, and proton magnetic resonance spectroscopy (H-MRS) may be more sensitive measures of underlying structural pathology.
ETIOLOGY AND IMMUNOPATHOGENESIS
Although the cause of MS remains uncertain, our understanding of the underlying mechanisms and pathology has grown enormously.
▪SPECIAL CLINICAL POINT: The best formulation of the pathogenesis of MS is that the disease is an autoimmune process that occurs in a genetically susceptible individual after an environmental exposure.
This hypothesis is supported by an extensive and diverse literature but is based on three seminal discoveries. The recognition by Rivers that the acute paralytic encephalitis that occasionally followed rabies and small pox vaccination was an autoimmune reaction to contaminating self-proteins led him to the discovery of experimental allergic encephalomyelitis (EAE), which has since been studied extensively as an animal model for MS. The discoveries that genes influence disease susceptibility and specifically that the human leukocyte antigen class II region on the short arm of chromosome 6 is associated with the risk of developing MS have led to a multitude of studies suggesting that polygenetic influences predispose certain individuals to acquiring MS. Finally, the observations made regarding latitudinal gradient, migration, and disease clustering have strongly suggested an environmental role in the disease. Although no specific microbial agent has stood the test of time, modern molecular immunology has provided numerous potential mechanisms through which numerous infections could mediate autoimmunity.
Genetics
Genetic susceptibility to MS has long been suspected, based on widely differing prevalence in different ethnic populations. Family studies have found that first-degree relatives of patients are at 20-fold increased risk of developing MS. Furthermore, monozygotic twins are more likely to be concordant for MS (20% to 40%) than dizygotic twins (3% to 4%). Nonbiologic first-degree (adopted) relatives are no more likely to develop MS than the population at large, which further supports the notion that familial clustering for MS is largely genetic in origin. However, unless one invokes incomplete penetrance of genes, the twin studies also suggest a strong environmental component because no more than 40% of monozygotic twins are concordant for MS. Widely different MS prevalence among similar high-susceptibility genetic groups that inhabit different environments (such as Anglo-Saxons in England versus in South Africa) presumably must be tied to this second, environmental element of disease etiology. Studies of candidate genes and whole-genome screens suggest that multiple weakly acting genes interact epistatically to determine risk toward MS, as suspected from epidemiologic studies. Recently, various single-nucleotide polymorphisms (SNPs) associated with a higher risk of developing MS were identified on the interleukin-2 receptor a gene, interleukin-7 receptor a gene, and confirmed in the HLA-DRA locus.
Immunology of Multiple Sclerosis
The inflammatory reaction in MS is of unknown origin. Although no infectious agent has been proved to be the cause of MS, it is hypothesized that many common viruses can trigger autoimmune-mediated demyelination in susceptible individuals through molecular mimicry (cross-reactivity between microbial proteins and myelin). In addition, there is evidence that quiescent autoreactive T cells that are present in healthy individuals may be activated through bystander activation by cytokine or polyclonal T-cell activation mediated by bacteria or viruses.
▪SPECIAL CLINICAL POINT: It remains uncertain how tissue injury (to myelin, oligodendroglia, axons, and neurons) occurs in MS, but the inflammatory events surrounding the vast majority of acute MS lesions seem to suggest a direct immune-mediated pathology.
The MS lesion resembles a delayed-type hypersensitivity (DTH) reaction, containing activated T cells, B cells, numerous mononuclear phagocytes, inflammatory cytokines, and adhesion molecules. Electron microscopy studies suggest the macrophage may play a major role in the direct stripping of myelin from axons, although this could be a secondary phagocytic response to excitotoxic injury of oligodendrocytes or neurons. There has been more emphasis recently on the role of innate immune cells in MS, specifically, dendritic cells and microglia. These cells seem to help facilitate the proinflammatory immune response in MS and direct autoreactive T-cell function within the CNS through antigen presentation. Therapies directed toward these cells could prove to be very beneficial.
Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. Neurology. 1996;46:907-911
DISEASE COURSE AND CLINICAL PATTERNS
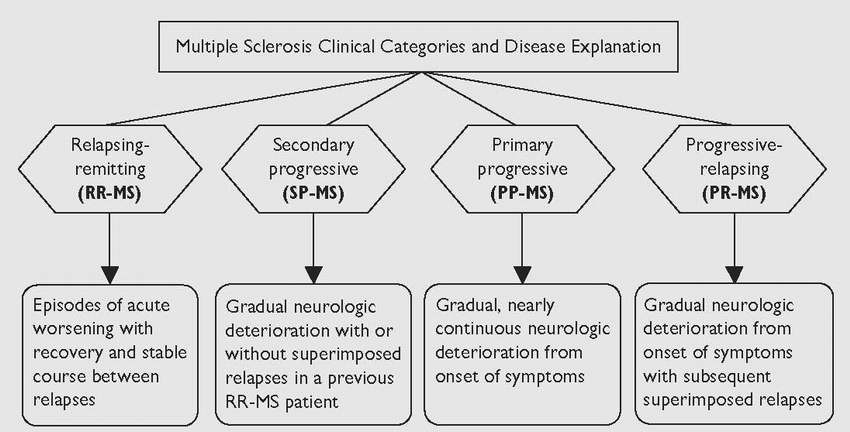
The clinical course of MS is divided into categories according to neurologic symptoms as they develop over time. A new classification for MS categories was developed by consensus among MS experts (Table 11.1).
Relapsing-Remitting Multiple Sclerosis
Relapsing-remitting MS (RR-MS) is the most common form in patients younger than age 40 years. Patients may develop focal neurologic symptoms and signs acutely or over a few days.
These exacerbations or attacks are remarkably unpredictable and heterogeneous in character, probably because they result from varying degrees of inflammation that can occur in any part of the brain or spinal cord.
▪SPECIAL CLINICAL POINT: Common presentations of multiple sclerosis include blurred or double vision, sensory symptoms (numbness, tingling, or pain), weakness, vertigo, or impaired balance.
A new symptom will commonly present over 24 to 72 hours, stabilize for a few days or weeks, and then improve spontaneously over 4 to 12 weeks. Subsequent new focal symptoms or signs typically follow the initial attack months or years later and again remit partially or completely. It is very common for old symptoms to persist or reoccur, especially in response to periods of stress such as infections or prolonged elevations of core body temperature. Over time, it becomes difficult to determine whether the symptom flare represents a new exacerbation or worsening symptoms referable to past disease. Recovery from relapses is often incomplete, and permanent disability can accumulate in a stepwise fashion at this stage of the disease.
Secondary Progressive Multiple Sclerosis
Secondary progressive MS (SP-MS) refers to the patient with an initial RR-MS course who then progressively worsens over months (at least 6) to years. Natural history studies have shown that with time most patients with RR-MS convert to SP-MS. This usually occurs after 10 to 20 years from onset or after the age of 40. A patient with SP-MS may still experience relapses but does not stabilize between relapses. The predominant clinical pattern is one of continued clinical worsening. As time passes, relapses become less discrete, and the pattern becomes one of continued worsening without relapses. Conversion to SP-MS is considered a poor prognostic sign because this stage of the disease is much more refractory to the presently available immunomodulatory therapies. Some patients with SP-MS spontaneously stabilize for considerable periods, although they only rarely recover after deficits have persisted for 6 months. The pathogenic mechanisms underlying conversion from RR-MS to SP-MS may relate to failure of remyelination and progressive axonal injury.
Primary Progressive Multiple Sclerosis
Primary progressive MS (PP-MS) accounts for approximately 10% to 15% of patients and is characterized by progressive worsening from the onset of symptoms without interposed relapses. Patients with PP-MS are more likely to be men and older than 40 years of age at symptom onset. This form of the disease often presents with progressive gait disorder as a result of leg weakness, spasticity, and impaired coordination. In cases of progressive neurologic dysfunction, it is extremely important to rule out structural pathology, infections, and hereditary and other neurodegenerative diseases. Patients with PP-MS have fewer gadolinium-enhancing brain lesions on MRI, less tissue inflammation on histopathologic assessment, and less cerebrospinal fluid (CSF) inflammation than typical for SP-MS; pathologic studies have suggested this form of the disease may represent a primary problem with the oligodendrocyte.
Progressive-Relapsing Multiple Sclerosis
According to the new classification, progressive-relapsing MS (PR-MS) refers to the rare patient with progressive disease from symptom onset, who subsequently experiences one or more relapses. In all likelihood, this is another form of SP-MS without clinically apparent relapses in the early stages of disease, and if considered separately, this group comprises only 6% or fewer of all patients with MS.
Disease patterns change over time, and it may be difficult at a given time to clearly categorize a patient’s disease. The problem is particularly difficult when a patient is converting from a purely relapsing-remitting disease course to a purely progressive disease course. This has been termed “relapsing progressive MS” by some and “transitional MS” by others, but these disease categories cannot be defined precisely by current methods. Observation for as long as 1 year may be required to categorize such a patient with confidence.
Clinically Isolated Syndromes and Prognosis
▪SPECIAL CLINICAL POINT: In the era of partially effective prophylactic MS therapies, there has been increased emphasis on making a diagnosis early in the course of the disease to initiate appropriate preventative treatment.
The use of MRI as a diagnostic tool is discussed later. T2-lesion burden at the time of first MS symptoms (optic neuritis, transverse myelitis, etc.) not only determines the likelihood of converting to clinically definite MS but is also predictive of future disability. For example, a patient with a single episode of optic neuritis and an abnormal brain MRI has a 50% to 80% risk of subsequently being diagnosed with MS in 5 to 10 years. The frequency of gadolinium-enhancing lesions correlates with the likelihood of having a clinical exacerbation and predicts future brain atrophy. However, because T2-weighted lesions lack specificity for tissue pathology and gadolinium enhancement is transient (2 to 8 weeks), Tl hypointense lesions (black holes) and brain atrophy may be a better measure of axonal loss and have been shown to correlate more strongly with both present and future disability. The presence of mild atrophy or persistent Tl black holes early in the course of MS should alert the physician to a potentially aggressive form of the disease.
OTHERVARIANTS OF MULTIPLE SCLEROSIS AND IMPORTANT MIMICKERS
Several other variants of MS have been described and are important to recognize.
Marburg Disease
An acute rapidly progressive form of MS, often called Marburg disease, is characterized by a person who develops acute or subacute progressive neurologic deterioration, leading to severe disability within days to months. The disease may progress steadily to a quadriplegic obtunded state with death as a result of intercurrent infection, aspiration, or respiratory failure from brainstem involvement. Postmortem studies have documented inflammation in the optic nerves, optic chiasm, cerebral hemispheres, and spinal cord. The pathology reveals a pronounced mononuclear cell infiltrate with severe axonal damage and tissue necrosis.
Neuromyelitis Optica
Neuromyelitis optica (NMO) or Devic disease refers to the patient who presents with both optic neuritis and transverse myelitis, occurring either simultaneously or separated by a few months to years. Several features differentiate this disease from classical MS, including older age of onset, higher female to male incidence (9:1), limited white matter lesions on a brain MRI, multilevel contiguous typically central spinal cord lesions (three or more vertebral levels), and severe disability and death as a result of respiratory failure in one third of all patients. NMO also seems to be more common than MS in non-Caucasians. Pathologically, the syndrome is variable with some lesions characterized by inflammation and demyelina-tion, but it is invariable with severe necrosis and many patients having cavitary lesions in the spinal cord. Those patients who survive the acute attack commonly follow a course with features indistinguishable from RR-MS but have a worse prognosis. An NMO serum bio-marker (NMO-IgG) has recently become commercially available, which can help differentiate this disease from MS. This distinction is of utmost importance since the therapeutic interventions are different for NMO and MS. While the NMO-IgG blood test is highly specific and the majority of clinically diagnosed NMO patients will be NMO-IgG positive, a negative blood test does not exclude the diagnosis.
Acute Disseminated Encephalomyelitis
Acute disseminated encephalomyelitis (ADEM) and its hyperacute form, acute necrotizing hemorrhagic encephalopathy (ANHE), are thought to be forms of immune-mediated inflammatory demyelination. They differ from MS in that they are typically monophasic, whereas MS is by definition multiphasic or chronically progressive. Patients with ADEM or ANHE usually present with fever, headache, meningeal signs, and altered consciousness, which are exceedingly rare in MS. Multiple reports of clinical and pathologic overlap have been published. Some authors have suggested that the MRI can be used to differentiate MS from ADEM, but no reliable clinical criteria to differentiate the two processes exist.
PRECIPITATING FACTORS
Exposure to viruses and bacteria has been associated with precipitating disease exacerbations. The risk of an exacerbation decreases during pregnancy, with the rate being decreased by approximately two thirds in the third trimester. The risk of an acute attack in the first 3 months postpartum is increased and has been estimated that 20% to 40% of postpartum patients with MS will have an exacerbation. The decreased relapse rate during pregnancy is probably related to a family of immunosuppressive hormones and Th2. Overall, pregnancy probably has little overall effect on the course of the disease, and therefore remains a realistic possibility for woman with MS. There is increasing evidence that vitamin D deficiency may play an important role in MS. Recent studies have suggested that higher serum vitamin D levels correspond to a decreased risk of developing MS. It is well established that vitamin D receptors are present within immune cells and may promote immune regulation. The anti-inflammatory cytokine, transforming growth factor-fij (TGF-Pj), is typically decreased in MS and increases upon vitamin D supplementation. This association further supports the contributions of environmental factors, especially since sunlight exposure provides a major source for vitamin D. This could also explain the unusual geographical distribution of MS.
Multiple sclerosis commonly affects the optic nerves and chiasm, and approximately 30% of patients present with visual symptoms. In acute optic neuritis, the patient experiences monocular loss of central vision and often has eye or brow pain, which worsens on lateral eye movement. The symptoms may present over a few hours to 7 days, with a few cases progressing over several weeks. Loss of visual acuity and color perception are often considerable. Most patients will recover significantly after 2 to 3 months, although continued improvement can occur as long as a year later; however, some patients sustain permanent damage and can become blind. Acuity is variably diminished in the affected eye. A central or centrocecal scotoma (marked enlargement of the blind spot to involve central vision) can be documented at the bedside with an Amsler grid, and red desatura-tion can be demonstrated with color plates or with a red-tipped hat pin. Using the swinging flashlight test in a darkened room, one can demonstrate a defect in the afferent pathway such that pupillary constriction in the affected eye is greater with contralateral than with direct light stimulus. A positive test reveals a relative afferent pupillary defect, sometimes called a Marcus Gunn pupil. Funduscopic examination is usually normal in acute retrobulbar neuritis. When the optic nerve is affected anteriorly, the disc may be congested and swollen, thus resembling papilledema. Several months after an optic neuritis, the disc often appears pale, especially at the temporal border, and this can provide evidence of a previous attack. Occasionally, optic nerve demyelination and axonal damage can manifest silently and is noticed only in the setting of other symptoms suggestive of MS.
TABLE 11.2 Initial Symptoms in Patients with Multiple Sclerosis
Symptom
Percentage
Sensory disturbance in one or
more limbs
33
Disturbance of balance and gait
18
Vision loss in one eye
17
Diplopia
13
Progressive weakness
10
Acute myelitis
6
Lhermitte”s symptom
3
Sensory disturbance in face
3
Pain
2
Paty DW, Poser CM. Clinical symptoms and signs of multiple
sclerosis. In: Poser CM, ed. The Diagnosis of Multiple Sclerosis.
NewYorlcThieme & Stratton; 1984:27.
TABLE 11.3 Common Symptoms and Signs in Patients with Multiple Sclerosis
Symptom
Sign
Comment
Visual blurring (central)
Diminished acuity (central)
Syndrome of optic neuritis seen usually early in disease
Vision loss/eye pain
Scotoma/deafferented pupil
Diplopia
Internuclear ophthalmoplegia
May be associated with nausea, vertigo, or other brainstem signs
Oscillopsia
Rarely other oculomotor weakness, flutter, or dysmetria
Loss of dexterity
Upper motor neuron signs often affecting legs early and arms later
Develops in many MS patients over time
Weakness
Tightness and pain
Shaking
Intention tremor, dysmetria, dysarthria, truncal or head titubation
Occurs in 30% of patients; may be the predominant manifestations in some patients
Imbalance
Paresthesias
Decreased vibration and position sense in legs > hands
Sensory symptoms are often
painful and distressing
Loss of sensation
Decreased fine sensation in hands
Bandlike disturbance
Sensory level
Falls
Wide-based gait
Gait disturbances are common in MS and can cause severe disability
Lack of coordination
Ataxic and unsteady gait
Inability to concentrate
or learn
Diminished concentration, processing speed, or verbal learning on neuropsychologic testing
May be subtle or have severe impact on patient and family; severe dementia in < 10% of patients
Easily distractible
Emotional lability
Episodic crying or laughing
Distressing to patient;generally not related to patients’ actual emotions
Depression
—
Commonly underrecognized or underestimated
Fatigue
—
Disabling in many patients with MS; does not correlate with severity of motor signs
Pain
—
Numerous etiologies (see text)
Urinary urgency, hesitancy, frequency, and incontinence
Requires urodynamic testing to fully characterize type of bladder dysfunction
A novel noninvasive eye scan called optical coherence tomography (OCT) is now being used to assess MS patients. The scan provides high-resolution quantifiable images of the retinal nerve fiber layer that have been shown in recent studies to reflect optic nerve damage even in asymptomatic eyes. OCT scans may prove to be a very important biomarker for disease monitoring and therapeutic effectiveness; however, since OCT scans are not specific for MS/optic nerve pathology, they are not considered a diagnostic test to the exclusion of a thorough eye exam.
Oculomotor Syndromes
Eye movement abnormalities are also extremely common in MS and include broken (saccadic) smooth pursuits, nystagmus, ocular dysmetria (overshooting target), isolated extraocular muscle palsies, and the classical internuclear ophthalmoplegia (INO).
▪SPECIAL CLINICAL POINT: An INO results from damage to the medial longitudinal fasciculus, and its presence in a young adult, particularly when bilateral, is highly suggestive of MS.
Only gold members can continue reading. Log In or Register to continue