Myoclonic–Astatic Epilepsy of Early Childhood – Definition, Course, Nosography, and Genetics
B. A. Neubauer*
A. Hahn*
H. Doose†
I. Tuxhorn‡
*Department of Neuropediatrics, Zentrum Kinderheilkunde und Jugendmedizin, University of Giessen, Giessen Germany
†Norddeutsches Epilepsie-Zentrum, Raisdorf, Germany;
‡Epilepsy Center Bethel, Klinik Mara, Bielefeld, Germany
Introduction
The first concept of myoclonic-astatic epilepsy (MAE) dates back to the 1960s when myoclonic epilepsies of early childhood were separated from infantile spasms and Lennox–Gastaut Syndrome (LGS). The prominent familial aggregation together with the characteristic seizure symptomatology dominated by myoclonic and myoclonic astatic seizures led Doose and co-workers to delineate MAE as a syndrome in its own right (1). Family studies of history and EEG suggested a genetic etiology. This notion of an idiopathic epilepsy was questioned and not accepted however because of the variable course and frequent deleterious course. Severe MAE was, therefore, categorized into the cryptogenic epilepsies by the ILAE Commission. Nevertheless, the discovery of SCN1A and SCN1B mutations in four unique or rare families with GEFS+ (generalized epilepsy febrile seizure plus) whose phenotype included MAE provide support for the genetic basis of some cases of MAE. Channel gene defects were identified, thereby proving the conceptual robustness of MAE as a genetic disorder. In its early delineation, MAE was mixed with other myoclonias of infancy including SMEI, BMEI, and other unclassifiable cases of myoclonic epilepsies. Further work by various authors have substantially disentangled the many phenotypes and genotypes of myoclonias in infancy from MAE. A more specific syndrome of primary generalized or genetic myoclonic astatic seizures have become evident. Presently modern therapy has significantly improved prognosis, although, in some cases, outcome can still be unpredictable.
Definition
Myoclonic astatic epilepsy, as a rule, occurs in children with an uneventful history. Like most other myoclonic epilepsies of early childhood, it clearly affects more boys than girls. The sex ratio is about 2.7:1 (2). If inclusion criteria only considers children older than 1 year, this ratio might even reach 3:1 (3). Epilepsy starts in 94% in the first 5 years. In 24%, onset of seizures occurs during the first year of life (2,4,5). The observed seizure symptomatology includes myoclonic, myoclonic-astatic, generalized tonic-clonic, tonic seizures with generalized onset, and a nonconvulsive status epilepticus formerly referred to as “status of minor seizures.”
Seizure Semiology
Table 11-1 summarizes the seizure symptomatology of the first 109 diagnosed cases (1,2,4,5). In about 60% of cases, epilepsy starts with febrile or afebrile generalized tonic-clonic convulsions. Alternating grand mal and hemi-grand mal are a possible presentation. Some days or a few weeks later, myoclonic and/or myoclonic-astatic seizures appear in abundance, frequently in combination with short absences. At first, this occurs predominantly after awakening. Infrequently sleep-related, tonic axial seizures may manifest during its long course and are not necessarily associated with an unfavorable prognosis. The same holds true for the rare partial seizures that eventually can be observed in MAE.
TABLE 11-1. Seizure Symptomatology in 109 MAE Cases | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
Myoclonic seizures consist of symmetric, mostly generalized jerks, accentuated in the arms and the shoulders and frequently associated with a simultaneous flexion of the head. The intensity of these seizures is variable and ranges from violent myoclonic jerks with sudden falls to abortive forms merely presenting as short irregular twitches of the face.
Myoclonic-astatic seizures are characterized by a loss of muscle tone preceded by a (short) myoclonia. In polygraphic recordings, the loss of muscle tone corresponds to a silent period in the electromyography (EMG) that is paralleled by the slow wave in the EEG following the spikes or polyspikes of the myoclonus (Fig. 11-1). Myoclonic seizures with and without a discernable myotonia frequently occur together or in series. The myoclonia and the following myotonia equally contribute to the characteristic myoclonic-astatic seizure (6,7) see Chapter 3 and 15 (8).
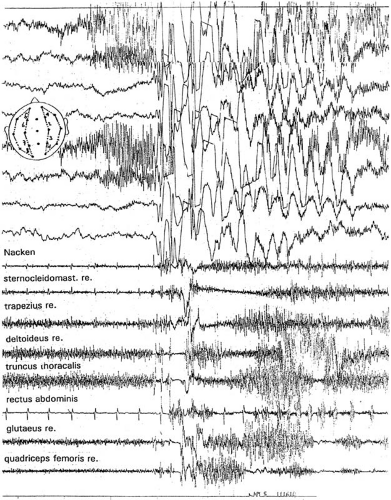 FIG. 11-1. Polygraphic record of a myoclonic-astatic seizure in MAE. The seizure was induced by startle. Initial spikes are associated with a short myoclonic jerk. The following slow wave is accompanied by a silent period in the deltoid, truncus thoracalis, gluteal and quadriceps femoris muscles. (Recorded in: Schweizerische Epilepsieklinik Zürich, 1992; from Doose H. EEG in childhood epilepsy–Initial presentation and long-term follow-up. Ist ed. Montrouge: John Libbey, 2003. With permission). |
Absences are seen in more than one-half of the children with MAE. Myoclonic and astatic seizures, when they come in series, are frequently accompanied by absences that are often combined with myoclonic jerks.
A peculiar type of nonconvulsive-status epilepticus (status of minor seizures) can be oberved in 36% (1,2,4) to 95% (3) of MAE patients. The characteristic clinical picture is a somnolent, stuporous child with subtle myoclonic seizures, frequently involving the face and the extremities. The child is unresponsive, drools, has slurred speech, or is even aphasic. This status may continue for days if not interrupted by adequate means.
Electroencephalogram
Background activity is of special interest in MAE. In cases beginning with GTCS, the EEG may stay entirely normal for weeks. However, in almost all instances, a rhythmic parietally accentuated 4–7 Hz activity develops early in the course (Fig. 11-2B). This rhythmic slowing of background activity was frequently questioned and falsely attributed to drowsiness. In patients with MAE (and other idiopathic generalized epilepsies of early childhood with myoclonic seizures), it represents a constant trait that is not related to the state of vigilance. This has been documented by EEG recordings of children who were kept attentive by displaying cartoons, etc. (8, p. 24). During the early stages, spikes, irregular spikes, and polyspikes may well be absent and appear only after some delay, starting during sleep.
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Myoclonic Status in Nonprogressive Encephalopathies
Myoclonic Status in Nonprogressive Encephalopathies
 Myoclonic Absences: The Seizure and The Syndrome
Myoclonic Absences: The Seizure and The Syndrome
 Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


