Myoclonic Seizures in the Context of Generalized Epilepsy with Febrile Seizures Plus (GEFS+)
Michel Baulac*
Isabelle Gourfinkel-An*
Stephanie Baulac†
Eric Leguern†
*Neurology/Epilepsy Department and Paris VI University, Hôpital Pitie-Salpetriere, Paris, France
†INSERM U 289, Hôpital Pitie-Salpetriere, Paris, France
‡Département de Génétique, Cytogénétique et Embryologie, Hôpital Pitie-Salpetriere, Paris, France
Introduction
The GEFS+ Familial Condition
Generalized epilepsy with febrile seizures plus (GEFS+) is a recently described familial condition (1) in which febrile seizures (FS) and epilepsy coexist in affected families. Indeed, in this heterogeneous familial context, some affected members have febrile seizures which are particular, called febrile seizures plus (FS+), because they persist beyond the classical limit of 6 years of age. They are often multiple in a given individual. On the other hand, some other family members may have typical FS, disappearing before the age of 6. Moreover, variable types of afebrile seizures are observed in affected individuals, most often generalized seizures, tonic-clonic (GTCS), absence, atonic, tonic seizures, including myoclonias and myoclonic seizures as well. Hemiconvulsives, temporal, and frontal lobe seizures have been also reported, extending the GEFS+ spectrum to partial seizures (1,2). These afebrile seizures may begin in childhood in association with the FS, this continuum between febrile and afebrile seizures being very characteristic (Fig. 8-1). However, afebrile seizures may also appear after a variable seizure-free period, or in subject without known previous history of FS.
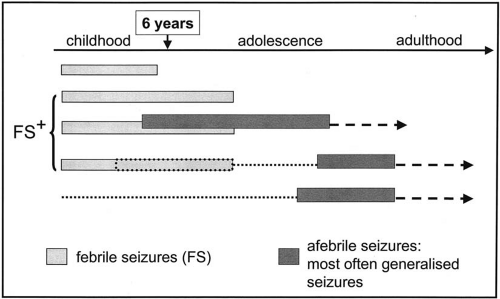 FIG. 8-1. Familial GEFS+ context. Temporal sequence of clinical expression of febrile seizures and afebrile seizures. |
Several types of seizures, including myoclonic seizures, can coexist in a given patient with more or less typical electroclinical features of idiopathic generalized epilepsies (Fig. 8-2). In some instances, they may constitute myoclono-astatic epilepsy (MAE) or severe myoclonic epilepsy in infancy (SMEI), although this latter syndrome has, thus far, been very rarely reported in the GEFS+ pedigrees. Finally, some electroclinical patterns that do not correspond to any of the well-established syndromes of the international classification can be encountered.
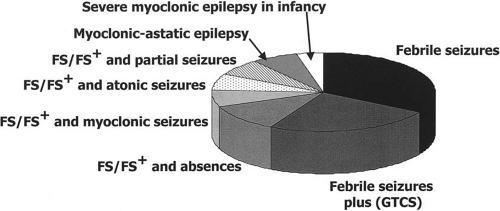 FIG. 8-2. GEFS+ phenotypic variability. Diversity of seizure types. (Adapted from Scheffer and Berkovic, Brain 1997.) |
Place of Myoclonic Seizures in the GEFS+ Spectrum
In the GEFS+ familial context, the spectrum of individual phenotypes includes all the generalized seizure types. Myoclonic seizures have been reported in patients with FS or FS+.
In one of their families, Singh et al. (3) describe two individuals: one had two FS at the age of 1.5 years and a brief period of frequent myoclonic seizures at the age of 2 during an intercurrent infection. He then became seizure-free off treatment at the age of 4. The other individual, evaluated at the age of 5, had FS, afebrile GTCS, and myoclonic seizures starting at the age of 1.
In another family, consistent with the GEFS+ phenotype (2), one individual only, aged 18 at the time of the study, presented myoclonic seizures. He had several FS, starting at the age of 1, including an episode of prolonged FS, and then, without free interval, he started to have GTCS accompanied by other seizure types. Myoclonic jerks were sporadic. Absence seizures were consistent with the description usually made in GEFS+, with rare episodes lasting longer than the typical absences. He became pharmacoresistant and had moderate intellectual impairment. EEG recordings showed a slow background and generalized spike-waves. Myoclonic jerks were not directly observed and polyspike waves were not recorded, however.
In spite of their well-established presence in the GEFS+ spectrum, myoclonic seizures account for a small proportion of the diverse seizure types. In a survey of ten families consistent with the GEFS+ phenotype, including 94 affected individuals, typical FS were reported in 35%, FS+ in 25%, FS or FS+ and absences in 7.5%, while FS or FS+ and myoclonic seizures were reported in only 3% of the patients (3).
Myoclonic seizures are also present as one of the seizure types of the syndromes that can be observed in GEFS+ families. MAE is the most frequent of these syndromes and ten of them are reported in a meta-analysis of ten families (3). However, eight out of these ten cases were observed in each of the eight small families included in this survey. The mean age of seizure onset was 1.5 years, all but one with fever. Four had explosive onset of multiple seizure types and developed a combination of myoclonic, absence, and GTCS seizures, myoclonic seizures occurring singly or in clusters.
SMEI, which is usually a sporadic condition, is a relatively rare phenotype in GEFS+ pedigrees and very few affected members have, thus far, been reported (4). Some studies, however, lead to the observation that FS and FS+ could be encountered more frequently than expected in SMEI relatives. Among 39 affected relatives of 12 SMEI probands, 14 relatives with FS, 7 with FS+, 2 with partial epilepsies, and single individuals with SMEI, MAE or Lennox–Gastaut syndrome (LGS) were identified (5). These findings suggested that SMEI could be the most severe condition of an extended GEFS+ spectrum.
Finally, juvenile myoclonic epilepsy (JME) seems to be a rare condition in GEFS+. One case was reported in the original pedigree leading to the initial delineation and description of the GEFS+ condition, but at the stage of molecular analysis it was found that this patient did not have the mutation that was present in most of the other affected members (6). Another very probable case is reported as the proband for a family consistent with the classification of GEFS+. He had myoclonic seizures without preceding FS, with a polyspike wave pattern (7).
The conclusion seems to be that myoclonic jerks are not a very frequent phenotype in GEFS+ families, although a few individuals have the characteristic sequence FS or FS+ and myoclonic seizures. The interest in myoclonias occurring in GEFS+ families, however, lies in all the genetic findings that have been made in this context and which may, to some extent, contribute to the pathophysiology of myoclonic seizures.
Genetics Findings in GEFS+
GEFS+ is inherited as an autosomal dominant trait with incomplete penetrance (70–80%); it is genetically heterogeneous. The first locus was mapped to the chromosome 19q13.1 and a mutation (Cys121Trp) in the SCN1B gene, encoding the β1 subunit of the neuronal voltage-gated sodium channel, was identified in an Australian family (8). This mutation altered the tertiary structure of the protein by disrupting a disulfuric bridge in the extracellular immunoglobulin-like domain of the auxiliary β1 subunit of the voltage-gated sodium channel. Using electrophysiological recordings from mammalian cell lines, Meadows et al. (9) reported that Cys121Trp mutation causes subtle effects on channel function and subcellular distribution that bias neurons toward hyperexcitabity and epileptogenesis. A few years later, only two other mutations in SCNIB were identified in GEFS+ families (10) and this gene has been excluded in the vast majority of families with GEFS+.
A second disease locus mapping to region 2q21-q33 has been reported (2,11). In the two French families studied, two different mutations (Arg1648His and Thr875Met) have been identified in the SCN1A gene. This gene encodes the α1 subunit of the neuronal voltage-gated sodium channel, which forms the pore of the channel (12). These two mutations are located in the transmembrane segment S4 of II and IV domains that are responsible for channel activation in response to the potential. Mutations in the SCN1A gene seem to be more frequently implicated in GEFS+ families as additional numerous point mutations in this gene have been later described (6,7,13,14). Mutations in SCN1A were also reported in patients (isolated cases or familial cases) with febrile seizures and partial epilepsy (15,16). The SCN2A gene, also located in 2q21-q33 and encoding the α2 subunit of the voltage-gated sodium channel, may be also implicated (17).
Electrophysiologic studies have demonstrated that mutations in the β1, α1, and α2 subunits interfere with the functional properties of the sodium channel (8,18,19,20,21,22).
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Myoclonic Status in Nonprogressive Encephalopathies
Myoclonic Status in Nonprogressive Encephalopathies
 Myoclonic Absences: The Seizure and The Syndrome
Myoclonic Absences: The Seizure and The Syndrome
 Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


