Myoclonic Status in Nonprogressive Encephalopathies
Bernardo Dalla Bernardina*
Elena Fontana*
Francesca Darra*
*Servizio Neuropsichiatria Infantile, Policlinico G.B. Rossi, Studi di Verona, Verona, Italy
Introduction
The existence in infants and young children, with a nonprogressive encephalopathy, of epilepsies characterized predominantly by myoclonic manifestations and having, in their natural history, a significant risk to develop more or less frequently a myoclonic status is well documented in the literature (1,2,3,4,5,6,7,8,9,10).
Nevertheless, the reports outlining the existence of an epileptic syndrome essentially characterized by the recurrence of long-lasting or subcontinuous myoclonic status in children with a nonprogressive encephalopathy are rare (11,12,13,14,15,16,17,18).
Several authors (17,18,19,20,21,22,23,24,25,26) describe a quite similar electroclinical picture in children with Angelman syndrome and in some children with 4p- syndrome (27), but few authors have stressed how, in some of these cases, the electroclinical picture was typically that of a “myoclonic status in nonprogressive encephalopathy” (12,13,14,15,27,28,29).
Following the results of our electroclinical longitudinal study of 51 cases, we describe this peculiar form of epileptic encephalopathy, recently proposed also in the scheme of the ILAE Task Force on Classification and Terminology (30) under the heading “Syndromes in development.” This form of epilepsy is characterized by recurrence of long-lasting myoclonic status appearing in infants and young children with a nonprogressive encephalopathy and having a variable poor prognosis.
Clinical and Electroencephalographic– Polygraphic Features
The 51 children studied include 34 females and 17 males with a sex ratio male:female of 1:2; their actual age is between 3 and 19 years (mean age 6 years). Five children (3 females and 2 males) died at the ages of 18, 24, and 30 months, and at 6 and 10 years, respectively. Familial antecedents of epilepsy were reported in 9 cases (17.6%).
The population can be divided into three etiological groups:
Genetic: Twenty-four subjects (47%; 17 females and 7 males). In 19 (37%), there was a deficit in chromosome 15q 11-q13, which is testimony to Angelman syndrome (13 females and 6 males). Two females presented a deletion of the short arm of chromosome 4 and the typical phenotype of the Wolf–Hirschhorn syndrome (currently known as 4p- syndrome). Two girls were affected by Rett syndrome and a Prader–Willi syndrome was diagnosed in one boy. No neuroradiologic abnormalities were present.
Fetal/neonatal anoxic injury: Eight subjects (15.6%; 2 females and 6 males). On neuroradiologic investigation, significant atrophic abnormalities were recognizable only in 3 subjects.
Unknown: Nineteen subjects (37%; 15 females and 4 males) were affected by a assessed symptomatic nonprogressive encephalopathy of which we were unable to define the etiology. The neuroradiologic picture [magnetic resonance imaging (MRI)] revealed a focal unilateral or bilateral micropolygyria in five subjects, a complete callosum agenesia with microcephalia in one subject, a slight colpocephaly with vermis hypoplasia and microcephalia in two sisters, and a partial callosum agenesia with bilateral hippocampal dysgenesis and vermis hypoplasia in another two sisters from another family. In 10 of 19 cases, therefore, the pathology appears to be constituted by a cortical dysplasia, in most cases, probably genetically determined.
The most constant clinical aspect at onset (38 patients, 75%) was that of an axial hypotonia of variable degree causing, in 23 cases (45%), a simple retardation in postural acquisitions; in 19 (37%), aposturality was associated with polymorphous abnormal movements presenting a picture of hypotonic “ataxic” cerebral palsy with a dystonic–dyskinetic syndrome and severe mental retardation.
Dysmorphisms evoking a probable genetic and/or malformative etiology were present at onset in 13 subjects (25%); slight microcephaly, reported at birth in 3, were present in 11 subjects (22%).
The average age of seizure onset was 10 months (range: 1 day–5 years). In 22 cases, the epilepsy onset was constituted by a myoclonic status characterized by very frequent during the day or subcontinuous “absences,” accompanied by periorbital and perioral myoclonias and rhythmic and arrhythmic jerks of distal muscles.
In the others, the initial seizures were mostly partial motor seizures, more or less typical brief myoclonic absences, massive myoclonias, and, more rarely, generalized or unilateral clonic seizures recurring in some patients only during cases of a febrile illness. Furthermore, the recurrence of brief massive startles was frequently reported at rest and during drowsiness.
The average age when myoclonic status was recognized was 14 months (range: 3 months–5 years). Because of severe mental retardation and continuous abnormal movements, both the paroxysmal attention disturbances (“absences”) and the myoclonias could remain hidden for several months in many cases. Considering the insidious appearance and probably the age at onset of myoclonias, the status, in many cases, may be unrecognized.
More frequently, myoclonias are subcontinuous but asynchronous in different muscles. In this case, their relationship with paroxysmal EEG activity is very difficult to appreciate; in this situation, the paroxysmal nature of the EEG pattern is also often difficult to recognize.
In some cases, the myoclonias are more obvious, rhythmically involving both arms and orofacial muscles. In other cases, the myoclonias are followed by a brief silent period, the result is a mixture of positive and negative phenomena. In some other cases, a negative myoclonus is predominant, which continuously fragments the voluntary movements and inhibits any fixed antigravity posturing.
Although the child is awake, the EEG is characterized by a slow activity with more or less easily recognizable paroxysmal abnormalities. These abnormalities consist of a relatively monomorphous subcontinuous, delta-theta activity (3–6 c/sec) varying in amplitude, and involving more or less asynchronously, the frontocentral regions. There are also brief sequences of rhythmic delta waves with superimposed spikes, achieving an unusual spike-and-wave, predominantly in parietooccipital regions, often elicited by eye closure (Fig. 5-1).
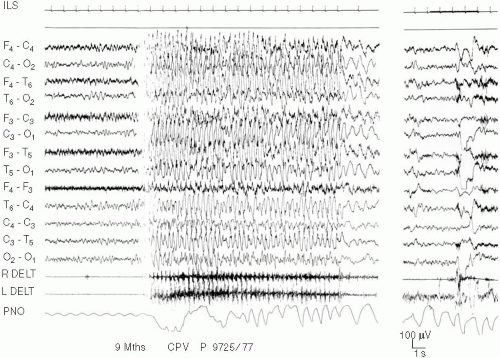 FIG. 5-1. A 9-month old girl with congenital microcephalia, bilateral frontocentral micropolygyria and callosum agenesia, massive axial hypotonia, and severe cognitive deficit presenting from the age of 5 months, with several daily brief myoclonic absences, evolving, a few months later, in long-lasting myoclonic status. See the relatively correct-for-age background activity and a myoclonic absence lasting 15 sec characterized by 3 per second diffuse spike-waves related with bilateral 3 per second rhythmic jerks inserted in a paroxysmally increased muscular tone. (R. Delt refers to right deltoid; ILS refers to intermittent light stimulation; PNO refers to pneumogram or respiration). |
The ictal manifestations are characterized by brief, more or less, diffuse bursts predominating in the central regions and slow spike and waves accompanied by bilateral, more or less rhythmic, myoclonias.
When myoclonias are rhythmic and synchronous in the two sides of the body, they are strictly related to EEG diffuse paroxysms in burst, achieving an ictal pattern, similar to that of a brief myoclonic absence. In this case, the ictal discharge is also characterized by a recognizable clinical “absence” (Fig. 5-1). More frequently the myoclonias are arrhythmic and become more easy to recognize during motor arrest, for example, during absence or drowsiness when all other abnormal movements disappear.
The electroclinical pattern of the status is often characterized by a fluctuation of the paroxysms. It is possible to observe, in fact, recurring bursts of spikes and waves or slow waves that are more or less diffuse, more or less synchronous, or asynchronous on both hemispheres. Between these bursts are inserted periods of variable duration without obvious paroxysmal discharges but with pseudorhythmic delta–theta activity of variable amplitude involving subcontinuously both central regions (Fig. 5-2).
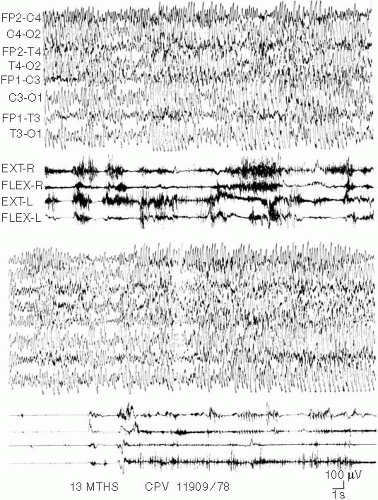 FIG. 5-2. A 13-month-old girl with ataxia, jerky movements, slight acquired microcephalia, cognitive impairment, speech absence, and frequent inadequate bursts of laughter. See the subcontinuous ample delta waves fluctuating in amplitude and diffusion, mainly asynchronous on the two hemispheres, related to subcontinuous more or less rhythmic jerks partially masked by the tonic component of the other abnormal movements. |
The proof that they are not separate ictal events recurring at a more or less high frequency, but a true status, is shown by the observation that, like theta activity, positive and negative myoclonias are also subcontinuous.
In some cases, for periods of variable duration, the status is more easily recognizable because it is characterized by a continuous diffuse, but asynchronous, sequence of spike and waves of great amplitude on both hemispheres. These are related to continuous rhythmic jerks, frequently followed by negative phenomena.
During drowsiness and slow sleep, spikes and waves become continuous to the point that the spindles are not recognizable. During stages II and III of the following nocturnal cycles, the paroxysm activation is minor and spindles are clearly represented; during slow sleep, the myoclonias vanish, reappearing briefly at arousal and eventually during rapid eye movement (REM) sleep when the diffuse discharges disappear in this phase. There is a continuous rhythmic theta activity mainly involving the vertex and rolandic regions. The same theta activity, strictly related to myoclonias, is transitorily observed during waking up.
Related posts:
 Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
Progressive Myoclonus Epilepsies: EPM1, EPM2A, EPM2B
 Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
Severe Myoclonic Epilepsy in Infancy: Dravet Syndrome
 Myoclonic Absences: The Seizure and The Syndrome
Myoclonic Absences: The Seizure and The Syndrome
 Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
Familial Juvenile Myoclonic Epilepsy
Ketogenic Diet in Patients with Dravet Syndrome and Myoclonic Epilepsies in Infancy and Early Childhood
 Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Treatment of Myoclonic Epilepsies of Childhood, Adolescence, and Adulthood
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


