2. Secondary hypokalemic periodic paralysis.
a. Thyrotoxic periodic paralysis occurs 70 times more often in men than in women, despite the increased prevalence of hyperthyroidism among women. In nearly all cases, the condition is sporadic and the attacks cease when thyroid function is normalized. Every patient with hypokalemic periodic paralysis needs screening for thyrotoxicosis. This condition is more common in patients of Asian, Hispanic American, and Amerind origin.
b. Periodic paralysis secondary to urinary or gastrointestinal potassium loss can result from primary hyperaldosteronism, excessive thiazide therapy, excessive mineralocorticoid therapy for Addison’s disease, renal tubular acidosis, the recovery phase of diabetic coma, sprue, laxative abuse, villous adenoma of the rectum, or prolonged gastrointestinal intubation or vomiting.
3. Hyperkalemic periodic paralysis produces episodic attacks of weakness accompanied by elevations in serum potassium level (up to 5 to 6 mmol/L). It can be associated with myotonia (inability to relax the muscle) or paramyotonia (muscle stiffness worsened by exercise or cold). It is inherited in an autosomal dominant manner. Attacks start in the first decade of life. Patients usually have brief periods of generalized weakness. Static weakness is rare. Sustained mild exercise may prevent attacks. Cardiac arrhythmias can occur.
a. The genetic abnormality is a mutation in the SCN4A (sodium channel) gene.
b. Needle EMG may detect myotonia, which supports the diagnosis.
4. Andersen’s syndrome (or Andersen–Tawil syndrome) is a triad of facial dysmorphism, long QT syndrome, and periodic paralysis. Short stature is often also present. Andersen syndrome is an autosomal dominantly inherited disease with a young age of onset and phenotypic variability. Fatal cardiac dysrhythmias may occur, making early recognition of this condition important. Mutations in the KCNJ2 gene, which codes for an inward-rectifying potassium channel, have been found in some patients.
5. Chloride channel mutations produce myotonia congenital with dominant and recessive forms (Thomsen and Becker, respectively), and with more myotonia than weakness. The dominant form manifests as painless muscle stiffness. Muscle stiffness is relieved after repeated exercise (warm-up), but it returns after rest. Cooling does not produce a significant change. These disorders result from missense mutations in the chloride channel gene CLCN1.
B. Prevention and therapeutic approach.
1. Primary hypokalemic periodic paralysis. Mild attacks may not require treatment. For attacks of general paralysis, oral potassium chloride can be used (0.25 mEq/kg), repeated every 30 minutes until the weakness is relieved. Muscle strength usually recovers within approximately 1 hour. IV potassium is not recommended because of the danger of cardiac arrhythmias and should be avoided.
For prevention of attacks, acetazolamide is the drug of choice, starting at 125 mg every other day, which can be increased to 250 mg three times a day. Side effects include increased incidence of nephrolithiasis, paresthesia, anorexia, and metallic taste. In severe cases, patients should eat a low-salt diet and be given the aldosterone antagonist spironolactone (100 mg twice a day) or triamterene (150 mg/day). Both drugs promote renal potassium retention.
2. Thyrotoxic periodic paralysis. Return to euthyroid status is curative. Propranolol (40 mg four times a day) and other β-adrenergic blocking agents may prevent attacks, possibly by suppressing the adrenergic overactivity induced by hyperthyroidism.
3. Hyperkalemic periodic paralysis.
a. Preventive measures consist of low-potassium diet, avoidance of fasting, and avoiding strenuous exercise. Slight exercise or ingestion of carbohydrates at the onset of weakness may prevent or abort attacks.
b. A thiazide diuretic, acetazolamide, or inhalation of a β-adrenergic agent (metaproterenol or salbutamol) may abort an attack. Dilantin (300 mg/day) can also be useful. For long-term preventive therapy, a thiazide diuretic or acetazolamide is recommended at the lowest possible dosage (hydrochlorothiazide, 25 mg every other day).
4. In myotonia congenita, mexiletine has been used. The starting dose is 150 mg by mouth twice a day, up to 1,200 mg/day. Mexiletine is contraindicated in the case of patients with second- and third-degree heart block; other cardiac arrhythmias can occur.
MUSCULAR DYSTROPHY
Muscular dystrophy is the term for inherited defects of cellular muscle structure, producing intrinsic muscle weakness. Some forms present at childbirth, others as late as the seventh decade. Family history, clinical examination, and temporal profile are necessary when considering a muscular dystrophy. The number of inherited dystrophies and the enormous variety of phenotypes prevent complete coverage in this forum. The following dystrophies will be discussed: X-linked dystrophinopathy (Duchenne and Becker muscular dystrophy), facioscapulohumeral dystrophy, myotonic, limb-girdle, and oculopharyngeal.
A. Natural history and prognosis.
1. Dystrophinopathy is an X-linked disorder caused by a mutation in the short arm, locus 21, of the X chromosome in the enormous gene that codes for the protein dystrophin. Dystrophin is a filamentous protein present in striated and cardiac muscle and other tissues. Although the role of dystrophin is not precisely known, anchoring and structural functions have been proposed for this protein.
In the most severe form of dystrophinopathy—Duchenne muscular dystrophy—almost no dystrophin is detected in skeletal muscle. In milder forms—phenotypically denominated as Becker muscular dystrophy—some muscle fibers express dystrophin, which may be structurally abnormal. Almost all patients with dystrophinopathy are male. The disease can be caused by spontaneous mutations, which are more common than in other genetic disorders, probably because of the large size of the gene. Approximately 70% of patients with Duchenne and Becker muscular dystrophy have detectable mutations on routine DNA testing of peripheral blood. Deletions of varying sizes can be found in approximately 65% of cases; 5% of patients have gene duplications. The remaining patients have a point mutation. The diagnosis in some cases of previously unreported point mutations is confirmed with dystrophin analysis at muscle biopsy.
a. Duchenne muscular dystrophy affects children early in life. Motor developmental delay is noticeable after the first year, but muscle necrosis and serum enzyme elevation can be found in neonates. Onset of walking may be delayed past 15 months of age. Signs are present before the age of 5 years. They include difficulties in running and climbing stairs.
Children have hyperlordosis with a prominent abdomen and calf pseudohypertrophy. Toe walking is common. To stand up from the floor, patients use their hands (Gower’s sign). Joint contractures of the iliotibial bands, hip flexors, and heel cords develop in most patients by 6 to 9 years of age. By the age of 10 years, many of these patients lose the ability to walk or stand and must use a wheelchair. By the midteens they lose upper-extremity function. Cognitive dysfunction occurs in 10% of cases. Although patients are living longer due to improved medical interventions, these patients have a shortened life span. Death is due to pulmonary infection, respiratory failure, or cardiomyopathy. Approximately 8% of female carriers have myopathy of the limb-girdle type. Female carriers may also have isolated cardiomyopathy.
Muscle biopsy specimens from patients with Duchenne muscular dystrophy have abnormal variations in fiber size, fiber splitting, central nuclei, and replacement by fat and fibrous tissues. The diagnosis of Duchenne’s muscular dystrophy on biopsy can be confirmed by an absence of dystrophin immunostaining.
An EMG obtained early in the course of the disease shows findings compatible with those of myopathy. In end stage, there are decreased numbers of muscle fibers and the tissue can even become inexcitable.
b. Becker’s muscular dystrophy is a milder variety of dystrophinopathy in terms of severity and molecular abnormality. The diagnosis has been typically defined as a patient who remains ambulatory after age 12. There is a wide range of phenotypic variability; some patients may live decades with mild symptoms, indistinguishable from those of limb-girdle dystrophy. All Becker-type patients are at risk for cardiomyopathy.
2. Facioscapulohumeral muscular dystrophy is an autosomal dominant disease that has high penetrance. It affects both men and women, most often presenting before 30 years of age. Ninety-five percent of patients have a deletion in a sequence of a 3.3-kb repetitive unit (known at D4Z4) in chromosome 4q35.
Clinically, facial muscles are affected early. Bell’s phenomenon (failure of eyelids to close completely when the patient is sleeping or blinking) and drooping of the lower lip are noticeable. Patients may be unable to whistle. Facioscapulohumeral muscular dystrophy also involves the trapezius, rhomboid, and serratus anterior scapular muscles. Scapular winging is noticed with forward arm movement because of serratus anterior weakness. Deltoid function and rotator cuff muscles are better preserved. Lower-extremity weakness is found later in the disease.
This disorder has wide phenotypic variability, even within the same family. Some patients remain ambulatory all their lives, whereas others progress to using a wheelchair. The heart is usually spared but cardiac monitoring is recommended.
3. Oculopharyngeal muscular dystrophy is an autosomal dominant disease of later onset. It is a GCG repeat disorder (8 to 13 repeats) involving the polyadenylate binding protein nuclear gene (PABPN) on chromosome 14. This dystrophy manifests as ptosis and progressive dysphagia. Muscle biopsy shows rimmed vacuoles in muscle biopsy specimens, and tubulofilamentous inclusions within the striated muscle cell nucleus. The differential diagnosis includes myasthenia gravis and mitochondrial myopathies (Kearns–Sayre syndrome).
4. Limb-girdle muscular dystrophy is a heterogeneous collection of both autosomal recessive and autosomal dominant disorders that affect pelvic and upper girdle muscles and spare the face. Some disorders present in childhood, others into late adulthood. There is CK, clinical phenotype and biopsy variability. In 2015, this remains a clinical diagnosis, but genetic testing with gene panels is improving.
5. Myotonic dystrophy is the most common muscular dystrophy among adults. Rather than being restricted to the skeletal muscle, it is a multisystemic, autosomal dominant disorder. It also involves the pancreas, gonads, thyroid, myocardium, and brain. Myotonic dystrophy is produced by a CTG trinucleotide repeat expansion in chromosome 19 (19q13.2–13.3) that codes for myotonin protein kinase, a ubiquitous enzyme related to protein phosphorylation. Patients are symptomatic when the CTG expansion is >80 repeats and the length of the repeat roughly correlates with severity of the disease. Clinical diagnosis is supported by the presence of myotonic discharges on EMG.
a. Muscle features. Weakness of facial muscles is typical. The face is hatched and thin with early frontal balding. Ptosis is present but is not as severe as in myasthenia gravis or Kearns–Sayre syndrome. Temporalis and masseter atrophy is a characteristic feature. Limb involvement is predominantly distal. Proximal limb muscles are usually preserved until the late stages. Myotonia, which is the delay of muscle relaxation after contraction, is present. Percussion of the thenar eminence or tongue can elicit myotonia. Patients may be unable to release their grip after a handshake.
b. Generalized features. Many patients have prominent systemic symptoms. Common abnormalities include cataracts, testicular atrophy, adult-onset diabetes mellitus, thyroid dysfunction, heart block, and arrhythmias. Hypersomnia and excessive daytime somnolence are reported and patients are often found to have mixed obstructive and central apneas. Because of the cardiorespiratory compromise, patients are susceptible to complications during surgery and anesthesia. Cognitive dysfunction, apathy, and lethargy are seen in more severely affected patients.
6. Myotonic dystrophy type 2 or proximal myotonic myopathy is an autosomal dominant disorder characterized by progressive weakness, myotonia, and cataracts. This is caused by a tetranucleotide repeat (CCTG) in the zinc finger protein 9 gene (ZNF9). These patients are a phenotypically milder form of myotonic dystrophy, with a later onset of symptoms.
7. Mitochondrial myopathies are a category of inherited diseases in which the genetic defect is either in the mitochondrial DNA or in a nuclear DNA gene that encodes for a protein involved in the mitochondrial respiratory chain. Because these defects all produce mitochondrial dysfunction, leading to decreased cellular energy production, patients manifest with symptoms related to oxidative stress. Seizures, encephalopathy, strokes, cardiomyopathy, muscle weakness (especially extraocular muscles), short stature, and hearing loss are common symptoms. The genetics underlying mitochondrial myopathies are complex. Mitochondrial DNA defects are maternally inherited and heteroplasmy (unequal distribution of affected mitochondria) may occur. Nuclear DNA genetic abnormalities follow Mendelian genetics.
Measurement of function of the components of the respiratory chain, muscle biopsy, MRI of affected organs, pedigree analysis, and genetic analysis of the mitochondrial genome can assist in making the diagnosis. Some of the more common phenotypes are mitochondrial encephalopathy with ragged red fibers, mitochondrial encephalopathy with lactic acidosis and stroke-like syndrome, chronic progressive external ophthalmoplegia, Kearns–Sayre syndrome, and Leigh’s disease.
B. Therapeutic approach.
1. Duchenne muscular dystrophy.
a. Family and patient education is important. A multidisciplinary clinic, which specializes in muscular dystrophy, can provide specialists and support to the patient and families.
Genetic counseling is recommended. Mothers and female siblings can be assessed for carrier status by assessment of serum CK or, if the patient has a documented genetic defect, through peripheral blood genetic analysis. However, negative results of mutation analysis in the mother do not rule out the risk of Duchenne muscular dystrophy affecting future pregnancies. Even with normal results of peripheral blood gene analysis, a mutation can be present in a percentage of oocytes (germline mosaicism).
b. Physical therapy is used to preserve mobility and to prevent early contractures. Passive range of motion exercises and adequate orthotics may prolong ambulation but do not stop disease progression.
Orthotics and splints can assist with managing contractures. All patients progress to wheelchair dependency: proper wheelchair assessments and fittings can lessen the development of scoliosis.
Patients with Duchenne muscular dystrophy are at high risk of side effects of general anesthesia. Succinylcholine or halothane should not be used because of the risk of episodes that resemble malignant hyperthermia. Adverse effects can be reduced with the use of nondepolarizing muscle relaxants.
c. Respiratory therapy. In later stages, noninvasive intermittent positive-pressure ventilation is useful, especially when patients retain carbon dioxide. Pulmonary exercises, use of a cough assist device and use of a respiratory vest may prevent pulmonary infections.
d. Medications. Prednisone (0.75 mg/kg/day) is recommended when children are still ambulatory to prolong this phase of life. Prednisone can improve neuromuscular strength after 1 month of treatment. The maximum effect is reached by 3 months. Side effects need to be addressed and close monitoring is needed.
e. Therapy for Becker muscular dystrophy follows principles similar to those of therapy for Duchenne disease, tailored to each patient’s level of strength.
2. In facioscapulohumeral muscular dystrophy and limb-girdle muscular dystrophy, treatment varies with individual patients. In patients with minimal symptoms, screening for cardiomyopathy and genetic counseling may be all that is needed. For patients with a foot-drop, ankle-foot orthoses may be prescribed. Physical therapy will be useful for range of motion, stretching, and gait assessment. When ambulation is impaired, a mobility evaluation can assess for wheelchair or motorized scooter needs.
3. In oculopharyngeal muscular dystrophy, blepharoplasty with resection of the levator palpebrae muscles may be needed. Dysphagia may be relieved with cricopharyngeal myotomy.
4. In myotonic dystrophy, only when myotonia is disabling, phenytoin (100 mg by mouth three times a day) can alleviate myotonia. In general, patients with myotonic dystrophy are not greatly concerned about the myotonia. The main goals are prevention and management of the systemic disease, especially cardiac arrhythmias.
5. Treatment of mitochondrial myopathies depends upon which organ systems are involved. Avoiding oxidative stress (hypoxia, ischemia, hypoglycemia, and infection) may help prevent exacerbations or worsening. A “mitochondrial cocktail” of antioxidants and vitamins has been developed to promote respiratory chain function and includes co-enzyme Q10, riboflavin, creatine, carnitine, B complex vitamins, and vitamins E and C.
METABOLIC MYOPATHY
Metabolic myopathies comprise a group of inherited disorders in which the defect is an alteration in the processing of carbohydrates or fats. Acid maltase deficiency, McArdle’s disease, and carnitine-O-palmitoyltransferase (CPT II) deficiency are reviewed (Table 52.2).
A. Natural history, prognosis, and treatment.
1. Acid maltase deficiency (Pompe’s disease) is an autosomal recessive glycogen storage disease caused by a deficiency in lysosomal α-glucosidase, which participates in the metabolism of glycogen into glucose. The infantile form is often fatal; there is also a less severe juvenile and adult-onset form. Infants with Pompe’s disease have hypotonia, macroglossia, cardiomegaly, and hepatomegaly. Adults suffer from slowly progressive myopathy with respiratory failure. Diaphragm and thigh adductor muscles are preferentially affected.
The cause of this disorder is a mutation in the acid alpha-glucosidase gene (GAA), located on chromosome 17q25. GAA enzyme activity evaluation is used for screening, but in borderline cases, molecular genetic testing is available.
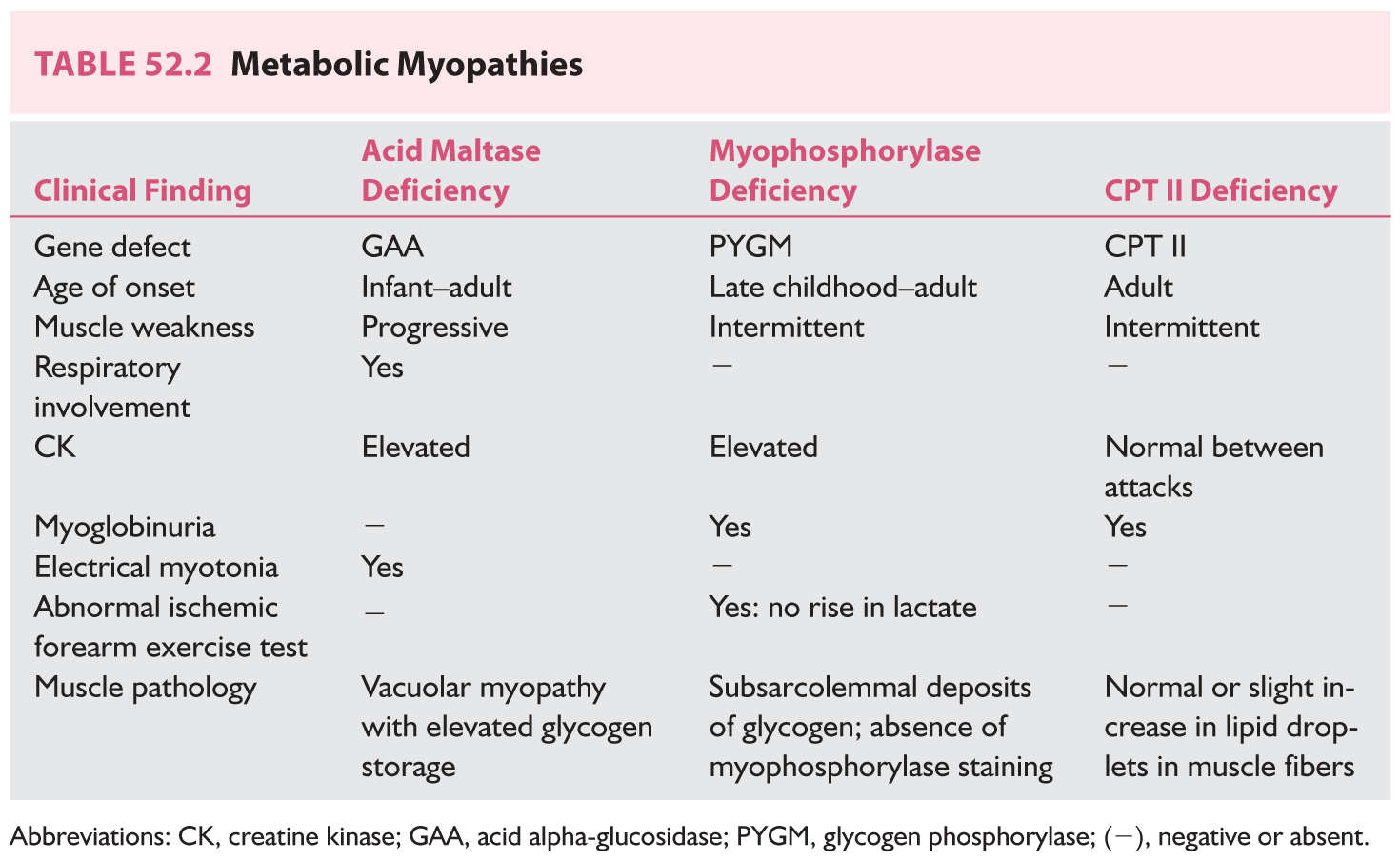
Treatment is now available for acid maltase deficiency, in the form of recombinant human α-glucosidase (Myozyme; Genzyme, Cambridge, MA, USA), given as repeated infusions. Enzyme replacement therapy has been clinically shown to lengthen the time before ventilator dependence in the infantile form. Adults have been documented to show increased strength and increased respiratory function.
2. McArdle’s disease, or myophosphorylase deficiency, affects children and adults and manifests with myalgia, fatigue, and muscle stiffness. Myoglobinuria and renal failure can develop. CK level is increased. EMG shows myopathic changes. The forearm ischemic exercise in affected patients shows no increase in venous lactate (unlike in normal controls). Muscle biopsy discloses subsarcolemmal deposits of glycogen. Immunohistochemistry staining of muscle biopsy tissue will show absence of myophosphorylase.
The disease is autosomal recessive, caused by homozygous or compound heterozygous mutations in the glycogen phosphorylase (PYGM) gene.
Prognosis is rather benign. Some patients can tolerate the deficits and learn to avoid brief, strong exercises that precipitate attacks. No definite treatment is available.
3. Carnitine-O-palmitoyltransferase II (CPT II) deficiency manifests in the adult patient with intermittent cramps, myalgia, and myoglobinuria. Renal failure, resulting from myoglobinuria, or respiratory failure may ensue. CK and EMG are normal between attacks. The symptoms are precipitated by intense exertion. The capacity to perform short exercise is not impaired. Fasting, exposure to cold, high fat intake, viral infections, and general anesthesia can precipitate rhabdomyolysis. Increased lipid content may be seen on histochemical staining on muscle biopsy, but is not always present. The disease is autosomal recessive. The diagnosis is made by sequencing and mutation analysis of the carnitine palmitoyltransferase II (CPT2) gene. Therapy includes avoidance of triggers (general anesthesia, prolonged exercise, cold exposure and prolonged fasting) and a diet high in carbohydrates and low in fats.
TOXIC MYOPATHY
Toxic myopathy is myopathy associated with either systemic disease or medication effect. Some medications produce direct muscle fiber necrosis, while others produce electrolyte imbalances with rhabdomyolysis. The most important types of toxic myopathy are necrotizing, autophagic, antimicrotubular, and steroid (Video 52.1). ![]()
A. Natural history, prognosis, and treatment.
1. Endocrine myopathy.
a. Thyrotoxic myopathy manifests as weakness and muscle wasting. Fatigue and heat intolerance are also present. Hypokalemic periodic paralysis (see Section A.2.a under Periodic Paralyses) and myasthenia gravis are associated with hyperthyroidism and should be included in the differential diagnosis. Treatment relies on correcting the hyperthyroid state; β-adrenergic blocking agents may be of help. Glucocorticoids should be used in thyroid storm to block the peripheral conversion of thyroxine to triiodothyronine.
b. Hypothyroid myopathy manifests as enlargement of muscles, weakness, painful cramps, myoedema, and slow-recovery reflexes. This disease is more common among women. Rhabdomyolysis or respiratory muscle involvement may be present. Serum level of CK may be elevated. The diagnosis is supported by abnormal results of thyroid function tests. Treatment is to restore the euthyroid state.
2. Toxic necrotizing myopathy. HMG-CoA reductase inhibitors (statins), used for cholesterol management, can cause a myopathy. Onset can be acute or insidious, often with myalgia, occasionally with myoglobinuria, and more often involving the proximal lower-extremity muscles. Patients with renal failure are especially predisposed. Elevated serum CK levels are common, and the EMG findings are abnormal. Muscle fiber necrosis with phagocytosis and small regenerating fibers are found on biopsy. When the medication is stopped, symptoms should resolve when the medication is stopped.
a. Asymptomatic elevations of CK level occur in about 1% of patients taking statins. These patients will need monitoring for muscle weakness.
b. Cyclosporine and tacrolimus have also been associated with toxic myopathy.
3. Autophagic myopathy can occur with chloroquine (and its derivatives) or amiodarone; it can be seen with systemic lupus erythematosus, scleroderma, and rheumatoid arthritis. The myopathy of chloroquine affects the proximal lower-extremity muscles and is usually not painful. The course is subacute or chronic. The heart can be affected. Elevation in CK level and myotonic potentials on the EMG can be found. Muscle biopsy shows vacuoles (lysosomes), which stain for acid phosphatase and contain debris and curvilinear structures (autophagic vacuoles). With amiodarone, severe proximal and distal weakness may occur in combination with distal sensory loss, tremor, and ataxia. The treatment is to discontinue the medication.
4. Antimicrotubular myopathy is produced by colchicine and vincristine. These drugs bind to nerve and muscle tubulin. The toxic etiology of this myopathy is often not recognized because of the insidious onset in patients who may have been taking colchicine for years. Concomitant axonal sensorimotor neuropathy occurs. Weakness resolves slowly. Because of the neuromyopathy, a mixed pattern of denervation and myopathy is seen on electrophysiologic studies. Muscle biopsy shows acid-phosphatase-positive autophagic vacuoles.
5. The effects of zidovudine can be indistinguishable from the myopathy of HIV infection. It is due to mitochondrial toxicity associated with this agent. CK levels are normal or mildly elevated. Differentiation from HIV myopathy can be difficult solely on a clinical basis. The muscle biopsy may show “ragged red fibers,” a sign of mitochondrial disease. Treatment consists of stopping the medication. Whereas myalgia (muscle pain) is usually relieved within weeks of discontinuing zidovudine, muscle weakness can persist for months.
6. Steroid myopathy is a type-2 fiber atrophy of muscles associated with long-term corticosteroid exposure. Doses of prednisone >30 mg/day carry the risk of myopathy. Fluorinated compounds (triamcinolone, betamethasone, and dexamethasone) have a greater risk. Patients have predominantly proximal muscle weakness and atrophy. Serum level of CK is usually normal. EMG findings are normal. Muscle biopsy shows type-2 fiber atrophy, especially type 2B (fast twitch glycolytic). Tapering to an alternate-day regimen, use of “steroid-sparing” drugs (e.g., azathioprine), use of nonfluorinated steroids, and exercise may reduce the incidence of this myopathy.
In polymyositis or dermatomyositis, clinical worsening in a patient being treated with steroids may represent either a progression of the primary disease or the onset of steroid myopathy. The decision to raise or lower the prednisone dose has to be made after evaluation of the patient’s muscle strength, mobility, CK levels, and medication changes in the preceding months.
Key Points
• Early and precise diagnosis of inflammatory myopathies is mandatory to initiate a correct treatment course, for the patient to get a good quality of life and a better prognosis.
• Care providers need to be aware of the myopathic consequences of statins, colchicine and chloroquine.
• Neurogenetics of muscle disorders has enormously expanded; new findings are leading to rational therapies.
• Care providers need to recognize the muscle complications of endocrine disorders, particularly those of thyroid, parathyroid, and adrenal glands.
• Specific treatments are now available for patients with acid maltase deficiency; therefore, this disease should be screened in all myopathic patients with diaphragmatic or thigh adductor weakness.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


