Chapter 84 Narcolepsy
Pathophysiology and Genetic Predisposition
Abstract
In its classical definition, narcolepsy is characterized by “excessive daytime sleepiness that typically is associated with cataplexy and other rapid eye movement (REM) sleep phenomena such as sleep paralysis and hypnagogic hallucinations”.1 Cataplexy, the sudden occurrence of muscle weakness in association with laughing, joking or anger, has long been considered the core pathognomonic symptom for the disorder.2–5 A broader definition of narcolepsy includes patients with sleepiness and abnormal REM sleep, such as sleep-onset REM periods (SOREMPs) during the multiple sleep latency test (MSLT), sleep paralysis, or hypnagogic hallucinations (narcolepsy without cataplexy). Disturbed nocturnal sleep is mentioned less often but is also common.2–6
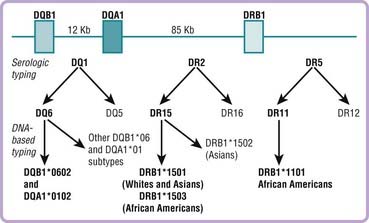
The definition of narcolepsy is being revised due to rapidly evolving scientific discoveries. Studies indicate that in most narcolepsy cases with cataplexy, and in fewer cases without cataplexy, a deficiency in the neuropeptide system, hypocretin is causal.7–12 A tight genetic association with the human leukocyte antigen (HLA) DQB1*0602 is also found in patients with cataplexy,13,14 suggesting an autoimmune mediation of the hypocretin cell loss. As a result, in the most recent revision of the International Classification of Sleep Disorders, narcolepsy with and without cataplexy have been separated (Table 84-1).1
Table 84-1 International Classification of Sleep Disorders
| CONDITION | DIAGNOSTIC CRITERIA | PATHOPHYSIOLOGY |
|---|---|---|
| Narcolepsy with cataplexy | Presence of definite cataplexy (and usually abnormal MSLT) | 95% with hypocretin deficiency and DQB1*0602 |
| Narcolepsy without cataplexy | ||
| Secondary narcolepsy | As for other forms of narcolepsy but with other conditions (e.g., neurologic) | With or without hypocretin deficiency; various disorders (see Table 84-4) |
| Idiopathic hypersomnia | No cataplexy, no SOREMPs during the MSLT | Unknown, likely heterogeneous |
For details, see International classification of sleep disorders. Diagnostic and coding manual. Chicago: American Academy of Sleep Medicine; 2005.
HLA, human leukocyte antigen; MSLT, multiple sleep latency test; REM, rapid eye movement; SL, sleep latency; SOREMP, sleep-onset REM period.
Narcolepsy treatments have also rapidly evolved, yet all remain based on symptoms. Available therapeutic options support the concept of a REM sleep and sleepiness duality in the symptoms of narcolepsy.15 Excessive daytime sleepiness is typically treated by amphetamine-like stimulants or modafinil. These compounds are effective in reducing daytime sleepiness but have little effect on cataplexy and abnormal REM sleep.4 Conversely, the most commonly used anticataplectic treatments, antidepressant drugs, alleviate cataplexy and other REM sleep abnormalities but have little effect on daytime sleepiness.4 A newly approved medication, gamma-hydroxybutyric acid (GHB or sodium oxybate), is effective in alleviating disturbed nocturnal sleep, cataplexy, and to a lesser extent daytime sleepiness. In this chapter, information regarding the pathophysiology and pharmacology of narcolepsy is reviewed. The clinical, diagnostic, and therapeutic aspects are discussed elsewhere16 and are not reviewed here.
Animal Models of Narcolepsy
In the last 20 years, narcolepsy research has been facilitated by the existence of a unique animal model, canine narcolepsy. Narcolepsy was first reported in two dogs by Knecht17 and Mitler18 in 1973. Early attempts to establish genetic transmission were unsuccessful, suggesting a nongenetic etiology in most cases of canine narcolepsy. In 1975, two narcoleptic Doberman pinschers were reported in a single litter.19 Subsequent breeding experiments found autosomal recessive transmission with full penetrance, allowing the establishment of a Doberman pinscher colony at Stanford University (Video 84-1 ). Familial canine narcolepsy was also reported in Labrador retrievers and dachshunds.19,20 Experiments indicate that animals heterozygous for the canine narcolepsy gene have subclinical abnormalities such as increased daytime sleepiness. In heterozygous animals, the administration of drugs that increase cholinergic and reduce monoaminergic transmissions (manipulations known to promote REM sleep) has been shown to induce cataplexy at specific developmental times.21
). Familial canine narcolepsy was also reported in Labrador retrievers and dachshunds.19,20 Experiments indicate that animals heterozygous for the canine narcolepsy gene have subclinical abnormalities such as increased daytime sleepiness. In heterozygous animals, the administration of drugs that increase cholinergic and reduce monoaminergic transmissions (manipulations known to promote REM sleep) has been shown to induce cataplexy at specific developmental times.21
The parallel between human and canine narcolepsy is striking. In MSLT-like procedures, narcoleptic canines have been shown to have short sleep and REM latencies.15 Twenty-four–hour recording studies show increased sleep fragmentation and proportionally more daytime sleep in narcoleptic than in control animals.22 Finally, as in human narcolepsy, sudden episodes of muscle weakness akin to cataplexy can be observed in association with strong positive emotions, most typically during the presentation of appetizing food or while at play (Fig. 84-1). These episodes usually last a few seconds and preferentially affect the hind legs, neck or face. Cataplexy may also escalate into complete muscle paralysis with abolition of tendon reflexes. During these episodes, the animal is conscious and most often able to visually track nearby movement (for video, see http://med.stanford.edu/school/Psychiatry/narcolepsy/moviedog.html). Polygraphic recording indicates a desynchronized, wakelike EEG pattern at the onset of cataplexy, followed by increased theta activity and genuine REM sleep in long-lasting episodes.23 A typical picture of narcoleptic canines in the midst of a cataplectic attack is presented in Figure 84-1.

The cause of autosomal recessive canine narcolepsy was identified through positional cloning.20,24 The pathology was found to be due to mutations in a receptor for the newly identified neuropeptide system hypocretin (hypocretin receptor-2). Three different mutations causing a complete dysfunction of the receptor were identified in Doberman pinscher, Labrador retriever, and dachshund pedigrees.20,24 Sporadic cases of canine narcolepsy were later shown to be associated with low CSF and almost absent brain hypocretin peptide content,25 as found in human narcolepsy.8,9
Several rodent models of narcolepsy are now also available. Chemelli and colleagues26 developed prepro-hypocretin knockout mice, reporting fragmented sleep and rapid transitions from wake into REM sleep. A reversible state of physical paralysis akin to cataplexy or sleep paralysis was also observed.26 In another model, a toxic transgene derived from an ataxin-3 human gene mutation, was driven by the hypocretin promoter, resulting in narcoleptic mice lacking hypocretin-containing cells.27 A rat model with partial hypocretin cell loss28 and mice lacking either of the two hypocretin receptors, hypocretin-1 and hypocretin-2, are now also available.29,30 In these models, only hypocretin receptor-2 knockout animals experience behavioral arrest episodes similar to cataplexy. Interestingly however, hypocretin receptor-1 knockout animals have fragmented sleep patterns but no behavioral arrest episodes.30 It is also suggested that hypocretin receptor-2 knockout animals are less affected than hypocretin peptide knockout animals, suggesting a role for HCRTR1 in increasing the severity of the phenotype.29
The use of these new rodent models, together with developments that make it possible to increase firing rate selectively in hypocretin cells through optogenic stimulation,31 is revolutionizing research in this area. Importantly, however, whereas it is clear that rodent hypocretin models all have a narcolepsy-like phenotype, it is difficult to differentiate cataplexy from REM sleep transitions or sleep paralysis at sleep onset. The link between behavioral arrest and positive emotions is unclear, and pharmacologic characterization has not been performed in narcoleptic rodents. Further, humans are more genetically diverse than inbred rodent lines and have distinct ecological niches, most likely explaining interspecies differences in the regulation of hypocretin.32 As an example, depriving mice of food for up to 31 hours has been shown to induce wakefulness, most likely because of the acute need to search for food, an effect abolished in mice lacking hypocretin.33 Yet, metabolically, mice have high demands and low metabolic reserves, unlike humans. Further, in contrast to these results, patients with hypocretin-related narcolepsy often use food restriction to stay awake, and they have binge-eating abnormalities, especially at night.34–36 Similarly, we found that the small decreases in food intake found in rodents with hypocretin deficiency could largely be explained by wake fragmentation in these models.37 Finally, as discussed later, pharmacologic responses can vary across species. As an example, the alpha2 antagonist yohimbine is a strong wake-promoting compound and anticataplectic agent in canines, yet it has only marginal if any effect in humans.15
The hypocretin system is ancient phylogenetically. It has been found in all mammals studied to date and in amphibians and fish but not in insects. Anatomically however, in zebrafish, a teleost vertebrate, hypocretin cells do not project heavily onto monoaminergic neurons in contrast to mammals. Further, hcrtr2-mutated zebrafish have disturbed sleep but not impaired daytime activity, suggesting a somewhat distinct physiologic role,38 although this is debated.39 These results suggest substantial interspecies differences in sleep and wake regulatory networks that are important to consider in the quest to understand the function of sleep.40
Pharmacology of Narcolepsy
Adrenergic Uptake Inhibition Mediates the Anticataplectic Effects of Antidepressants
In the past, the most commonly prescribed anticataplectic agents were tricyclic antidepressants. More recently, more selective monoaminergic reuptake inhibitors have become available. These compounds have a complex pharmacologic profile that includes monoamine (serotonin, norepinephrine, epinephrine, and dopamine) uptake inhibition and, for older tricyclic antidepressants, cholinergic, histaminic, and alpha-adrenergic blocking effects.15,41,42
Cataplexy is difficult to quantify in humans (because it is unpredictable) and mouse models (because it is difficult to differentiate from REM sleep or sleep paralysis), but this symptom can be easily measured in narcoleptic canines using a simple behavioral tool, the food-elicited cataplexy test. In this test, pieces of dog food are lined up on the floor and the animal is released into the room.15 A normal animal will complete the test in less than 10 seconds, whereas narcoleptic subjects, excited by the food, exhibit multiple partial or complete attacks that can be recorded in number and duration. This test has been used to pharmacologically dissect the mode of action of currently prescribed anticataplectic agents.15
In narcoleptic canines, results have shown that inhibition of adrenergic but not dopaminergic or serotoninergic uptake or other properties is critical to explain therapeutic efficacy for antidepressant compounds.41,42 This observation fits well with available human pharmacologic data15 (Box 84-1). Indeed, protriptyline, desipramine, viloxazine, and atomoxetine, four adrenergic-specific uptake blockers with no effect on serotonin transmission, are effective and potent anticataplectic agents (see Box 84-1). In contrast, escitalopram, zimelidine, fluoxetine, and other selective serotonin reuptake inhibitors (SSRIs) are either inactive or only active on cataplexy at relatively high doses, an effect probably mediated by the weak adrenergic uptake effects of these compounds and their metabolites in several cases.41 Alternatively, it may be that in humans, blocking the serotonin reuptake site has minor therapeutic effects on cataplexy in contrast to canines, because of increased genetic diversity in humans or species differences.
Box 84-1
For details, see references 15, 40, 41, and 45–48.
Pharmacologic Properties of Commonly Prescribed Treatments
The observation that adrenergic uptake blockers are anticataplectic agents correlates well with the potent inhibitory effects of these compounds on REM sleep. It is well established that adrenergic transmission is reduced during REM sleep.43 Firing rate in the locus coeruleus has also been shown to decrease during cataplexy in narcoleptic canines.44 Adrenergic uptake blockers might thus increase activity in adrenergic projection sites involved in REM sleep regulation. This effect would reverse the effect of decreased locus coeruleus impulse flow normally occurring during natural REM sleep. The fact that serotoninergic uptake blockers, also known to have inhibitory effects on REM sleep, have less or no effect on cataplexy is more surprising. Like adrenergic cells of the locus coeruleus, serotoninergic cells of the raphe nuclei dramatically decrease their activity during REM sleep.43 This discrepancy could be explained by a preferential effect of serotoninergic projections on REM sleep features other than atonia, for example in the control of eye movements. In this model, adrenergic projections may be more important than serotoninergic transmission in the regulation of REM sleep atonia and thus cataplexy.41 In favor of this hypothesis, one experiment has shown that serotoninergic activity does not decrease during cataplexy in narcoleptic canines,45 in contrast with locus coeruleus activity.44
Increased Dopaminergic Transmission Mediates the Wake-Promoting Effects of Currently Prescribed Stimulant Compounds
Commonly prescribed stimulant compounds include amphetamine-like drugs, such as dextroamphetamine, methamphetamine, methylphenidate, pemoline, and modafinil (see Box 84-1). Like tricyclic antidepressants, amphetamine-like drugs are nonspecific pharmacologically. Their main effect is to globally increase monoaminergic transmission by stimulating monoamine release and blocking monoamine reuptake. Abuse and dose escalation can occur with amphetamine, especially in patients without cataplexy. Less abuse is reported with methylphenidate, and modafinil is not believed to be addictive.
Studies have shown that the wake-promoting effect of these compounds is secondary to stimulation of dopamine release and inhibition of reuptake.46,47 The mode of action of modafinil is debated, but this compound also selectively inhibits dopamine uptake.48 All these compounds are ineffective in dopamine transporter knockout mice, suggesting a primary mediation of wake promotion via dopaminergic systems.47 Interestingly, compounds selective for dopaminergic transmission have no effect on cataplexy, whereas amphetamine-like compounds with combined dopaminergic and adrenergic effects have some anticataplectic properties at high doses.41,49 Adrenergic effects of amphetamine-like stimulants also correlate with the respective effects of these compounds on normal REM sleep.15,49 Dopaminergic-specific uptake blockers have little effect on REM sleep when compared to adrenergic or serotoninergic compounds.15 The most important effects of dopaminergic uptake blockers are to reduce total sleep time and slow-wave sleep.50 This preferential effect of dopaminergic uptake blockers on non-REM (NREM) sleep correlates with electrophysiologic data. As opposed to adrenergic or serotoninergic neurons, the firing rate of dopaminergic neurons is known to remain relatively constant during REM sleep.51,52
Interestingly, studies in humans and narcoleptic canines have shown that large doses of stimulants are needed to polygraphically normalize narcoleptic subjects. In our narcoleptic Doberman population, doses equivalent to 60 mg/day were needed to reduce daytime sleep to control levels.22 In both control and narcoleptic animals, however, the dose-response curves for modafinil or amphetamine were parallel. This result suggests that there is no difference in sensitivity to stimulants in narcoleptic animals but rather that higher doses are needed in narcoleptic animals because of their extreme baseline sleepiness.22 However, one study has suggested increased wake-promoting effects of modafinil in rodents with hypocretin deficiency.53
Sodium Oxybate (Gamma-Hydroxybutyrate)
Sodium oxybate, also called gamma-hydroxybutyrate (GHB), is a sedative anesthetic compound known to increase slow-wave sleep and, to a lesser extent, REM sleep.15 Abuse in the context of rave parties has been reported, and prescription of the compound is highly regulated, with centralized distribution. Because slow-wave sleep is associated with growth hormone (GH) release, GHB also induces GH release and has been abused by athletes. When administered at night, it consolidates sleep and improves daytime functioning. Because of its short half-life (~30 min), it must be administered twice a night. Interestingly, cataplexy and daytime alertness also improve over time, sometimes producing a full therapeutic effect only after several months of treatment and dose adjustments.16
The mode of action of GHB on sleep and narcolepsy is unclear. GHB has a major effect on dopamine transmission, reducing firing rate and raising brain content of dopamine.15,54 Other effects on opioid, glutamatergic, and cholinergic transmission have been reported.54 Specific GHB receptors have been identified, but the compound is also a GABAB agonist.54 Most studies to date suggest that the sedative-hypnotic effect is mediated via GABAB agonist activity.54,55 Whether this effect also mediates the anticataplectic effects after long-term administration is unknown.
Other Known Modulators of Narcolepsy Symptoms
The effects of more than 200 compounds with various modes of action have been examined in human patients and narcoleptic canines.15 In almost all cases except a few, similar effects were found in humans and canines.15 Almost all the effects have been reported for monoaminergic and cholinergic compounds. With cataplexy being easier to study than sleep in canines, most studies have also focused on cataplexy rather than sleepiness. For cataplexy, the findings were generally consistent with pharmacologic studies of REM sleep. As is the case for REM sleep, the regulation of cataplexy is modulated positively by cholinergic systems and negatively by monoaminergic tone.15 Muscarinic M2 or M3 receptors mediate the cholinergic effects, and monoaminergic effects are mostly modulated by postsynaptic adrenergic alpha1 receptors and presynaptic D2 or D3 autoreceptors.15
A number of studies have shown abnormal cholinergic and monoaminergic receptor density and neurotransmitter levels in brain and CSF samples human or canine narcolepsy.15,56–68 Local injection studies in selected brain areas of narcoleptic canines have also shown functional relevance for some of these abnormalities.69–71 As a result, cholinergic hypersensitivity, dopaminergic abnormalities, and decreased histaminergic tone are likely to be critical downstream mediators of the expression of the narcolepsy symptomatology.68–71 The cholinergic and monoaminergic imbalances observed in narcolepsy are best illustrated by the finding that in asymptomatic animals heterozygous for the hcrtr2 mutation, a combination of cholinergic agonists with an alpha1 blocker or a D2 or D3 agonist can trigger cataplexy.21 A possible application of these findings is illustrated by the recent development of histaminergic H3 antagonists, drugs that are known to stimulate histamine release via the H3 receptor, as novel wake-promoting stimulants for the treatment of narcolepsy.72
Hypocretin and Involvement in Narcolepsy
Anatomy and Physiology of the Hypocretin Neuropeptide System
The hypocretin/orexin system was discovered almost simultaneously by two groups of scientists, hence the two conflicting names. De Lecea and colleagues first isolated the prepro-hypocretin transcript (clone 35) and suggested the existence of two resulting processed neuropeptides sharing extensive homology with each other and weak homology with secretin.73 These neuropeptides were called Hypocretin 1 and Hypocretin 2, to indicate hypothalamic peptides of the incretin family. Using cell lines expressing various orphan G protein–coupled receptors (GPCR), Sakurai and coworkers screened tissue extracts for GPCR agonist activity.74 Two mature peptides stimulating the orphan HFGAN72 GPCR cell line were isolated and called Orexin-A and B.74 The name was chosen from Greek orexis (“appetite”) based on associated studies suggesting stimulation of appetite after central administration. A second receptor with high homology with HFGAN72 was also identified by homology. The two G protein–coupled receptors were initially called orexin receptor 1 and 274 but their official names are HCRTR1 and HCRTR2 in genetic databases. HCRTR1 (OxR1) binds hypocretin-1 (OxA) selectively, whereas HCRTR2 (OxR2) binds both hypocretin-1 (OxA) and hypocretin-2 (OxB) with similar affinity.74 The two receptors mostly couple with Gq and stimulate cellular activity in most cell types.75–77
Only a few thousand cell bodies containing these two peptides are found in the entire brain, all within the perifornical area of the posterior hypothalamus. Contrasting with this discrete perikaria localization, the neurons project widely in the central nervous system.75–77 Limbic system areas (including amygdala and nucleus accumbens), monoaminergic cell groups (adrenergic locus coeruleus, serotoninergic raphe nuclei, dopaminergic ventral tegmental area and substantia nigra, histaminergic tuberomammillary nuclei), intrahypothalmic nuclei, and various periventricular organs are densely innervated.75–77 Hypocretin immunoreactive varicose terminals are also seen in the cerebral cortex, spinal cord, and thalamus. The strongest extrahypothalamic prepro-hypocretin immunoreactive projection is found in the locus coeruleus. Hypocretin has also been suggested to be present in selected peripheral tissue (testis, gut) at very low levels of expression.
Hypocretin Deficiency and Human Narcolepsy and Cataplexy
As expected from the observation that most cases of human narcolepsy are sporadic and not fully genetic, as in dogs or mice, an extensive genetic screening study did not identify numerous prepro-hypocretin, hcrtr1, or hcrtr2 mutations in human narcolepsy.8,78,79 Surprisingly, even familial cases of narcolepsy (some of which were HLA-DQB1*0602 negative) did not have any hypocretin mutations, suggesting further heterogeneity in genetic cases.8 Rather, only a single case with a signal peptide mutation of the prepro-hypocretin gene was identified. This case has an extremely early onset (6 months), severe narcolepsy and cataplexy, DQB1*0602 negativity, and undetectable levels of hypocretin-1 in cerebrospinal fluid (CSF).8 This observation indicates that hypocretin system gene mutations can cause narcolepsy, as in animal models.
Following on the cloning of the canine narcolepsy gene, we have also found that most patients who have sporadic HLA-DQB1*0602–positive narcolepsy with cataplexy have undetectable hypocretin-1 levels in the CSF.7–1280 Follow-up neuropathologic studies in 10 narcoleptic patients also indicated dramatic loss of hypocretin-1, hypocretin-2, and prepro-hypocretin mRNA in the brain and hypothalamus of narcoleptic patients (Fig. 84-2).8,9 These subjects have no hypocretin gene mutations and a peripubertal or postpubertal disease onset81 as opposed to a 6-month onset in patients with a prepro-hypocretin mutation.8 Together with the tight HLA association,13,14 a likely pathophysiologic mechanism in most narcolepsy patients could thus involve an autoimmune alteration of hypocretin-containing cells in the CNS.
Hypocretin Transmission in Sleep Regulation
The potential role of hypocretin in the regulation of normal sleep is only beginning to emerge. Central (intracerebroventricular or local injections) but not peripheral administration of hypocretin-1 stimulates wakefulness and reduces REM sleep. Hypocretin antagonists promote NREM and REM sleep, including in humans. In rats and monkeys, cisternal CSF hypocretin-1 fluctuates, with maximal levels at the end of the active period (night in rodents) and minimum levels at the end of the inactive period (amplitude is 40%).82 Using in vivo dialysis, a similar profile is observed in rat brain tissue extracellular fluid.83 In diurnal, wake-consolidated squirrel monkeys, cisternal hypocretin-1 levels also peak in the late evening, around bedtime (amplitude ≥40%).84 These results suggest that hypocretin may be important to promote wakefulness in the evening in humans. In this model, hypocretin would oppose the sleep debt that has accumulated since early morning, allowing a constant level of wakefulness through the day.84 Additional studies suggest that diurnal fluctuations in hypocretin release are driven both directly by the circadian clock and indirectly by the increased sleep debt.32 It is also still uncertain at this stage how hypocretin release and activity fluctuates across the various sleep stages (REM versus NREM). In lumbar CSF, hypocretin-1 only has limited diurnal fluctuation (10%), suggesting a dampening and delay of changes when reaching the lumbar sack (minimal levels in morning).85,86 This finding is important practically because time of CSF collection has no significant effect for purposes of diagnosing narcolepsy.10,87
The most dense reported hypocretin projections are to the monoaminergic cell groups of the locus coeruleus (norepinephrine), substantia nigra and ventral tegmental area (dopamine), raphe magnus (serotonin), and tuberomammillary (histamine) neurons. Dopamine and histamine cell groups have one of the highest densities of hypocretin-2 receptors88 and may be especially important.75–7789 Increased dopamine level in the amygdala is one of the most consistent neurochemical abnormalities reported in canine narcolepsy.56,58 Decreased histamine levels are also observed in the brain of narcoleptic canines.68 In vivo dialysis studies have indicated a critical role for the dopamine mesolimbic and mesocortical system in regulating alertness and triggering cataplexy by emotions.
Histaminergic transmission has long been recognized as a critical wake-promoting neurotransmitter.90 Dopamine and histaminergic projections may thus be centrally involved in controlling both cataplexy and alertness. Similar to REM sleep, cataplexy is controlled by pontine cholinergic REM-on cells and aminergic locus REM-off cells.15 Removing hypocretin-excitatory projections to monoamine cell groups could decrease monoaminergic tone and produce a cholinergic-aminergic imbalance consistent with sleepiness and abnormal REM sleep in narcolepsy. Hypocretin projections to the basal forebrain area, an area with cholinergic hypersensitivity in narcoleptic animals, are also likely to be involved.69,70 It is also likely that hypocretin is critical to the integration of sleep regulation with metabolic status, although the importance of this effect could depend on the species.
Genetic Aspects of Human Narcolepsy
Familial Aspects of Human Narcolepsy
The familial occurrence of narcolepsy and cataplexy was first reported in 1877 by Westphal.91 Since then, numerous case reports have appeared in the literature. Until recently, narcolepsy was considered a familial disorder. More recent studies have shown that earlier reports were often confounded by unrecognized obstructive sleep apneas. One frequently cited publication by Krabbe and Magnussen92 reports that “narcoleptic” (obese) relatives of a narcoleptic proband would frequently fall asleep while playing cards, falling forward on the table, snoring loudly and becoming cyanotic during the episodes. In more recent studies, the risk of a first-degree relative to develop narcolepsy and cataplexy has been shown to be only 1% to 2% (see reference 93 for review). A larger portion of relatives (4% to 5%) may have isolated daytime sleepiness, when other causes of daytime sleepiness have been excluded.93 These figures are important to keep in mind because they are helpful in reassuring patients regarding the risk to their children and relatives. A 1% to 2% risk is 10- to 40-fold higher than in the general population but remains manageable. A 4% to 5% risk for daytime sleepiness is not negligible, but similar values have been reported for excessive daytime sleepiness in the general population independent of narcolepsy.94–96

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


