Figure 35.1. Three types of “seizures” in the newborn: “electrographic only,” “electroclinical,” and “clinical only.”
SIGNIFICANCE OF NEONATAL SEIZURES
Incidence
The incidence of seizures in the first 28 days of life, one of the highest risk periods for seizures in humans, ranges between 1% and 5%. Depending on the methodology used, seizures occur at a rate of 0.95 to 5.5 per 1000 neonates (1–6), most within the 1st week of life (3). Unfortunately, most of these studies relied on clinical findings to identify seizures, which may underestimate the actual incidence. Incidence varies with specific risk factors. Lanska et al. (3) reported the incidence of seizures in all neonates to be 3.5 per 1000, but 57.5 per 1000 in very-low-birth-weight (<1500 g) infants, 4.4 per 1000 in low-birth-weight (1500 to 2499 g) infants, and 2.8 per 1000 in normal-birth-weight (2500 to 3999 g) infants. Scher et al. (7,8) described seizures in 3.9% of neonates younger than 30 weeks of conceptional age and in 1.5% of those older than 30 conceptional weeks.
The human newborn is especially vulnerable to a wide range of toxic or metabolic conditions. It is now well established that the neonatal brain itself may be especially prone to seizures when injured. One suspected mechanism of enhanced seizure susceptibility in the newborn is the relative imbalance between inhibition and excitation. Compared with more mature brains, the neonatal brain exhibits delayed maturation of inhibitory circuits and precocious maturation of excitatory circuits (9). According to studies in the neonatal rat, γ-aminobutyric acid (GABA)—the major inhibitory neurotransmitter in the mature brain—may exert paradoxically excitatory effects in early CNS development (10,11).
A developmentally dependent cation chloride cotransporter channel (KCC2)—which extrudes chloride into the extracellular space—does not reach mature levels in the rat hippocampus until after the third postnatal week (Fig. 35.2). Instead, early in development, the NKCC1 transporter predominates and actively transports chloride into the neuron. Thus, when the ligand-dependent GABAA receptor is activated in the immature rat, extracellular chloride follows its electrochemical gradient out of the neuron resulting in depolarization. After the appearance of KCC2, the intracellular concentration of chloride is kept low, and activation of the GABAA receptor allows chloride to run along its electrochemical gradient into the neuron. This leads to hyperpolarization and allows for the inhibitory action of the receptor (12,13). Current electrophysiologic evidence suggests that this excitatory-to-inhibitory switch in the rat hippocampus is complete by postnatal day 14 (14,15), an age that may reflect the developmental state of a human toddler.
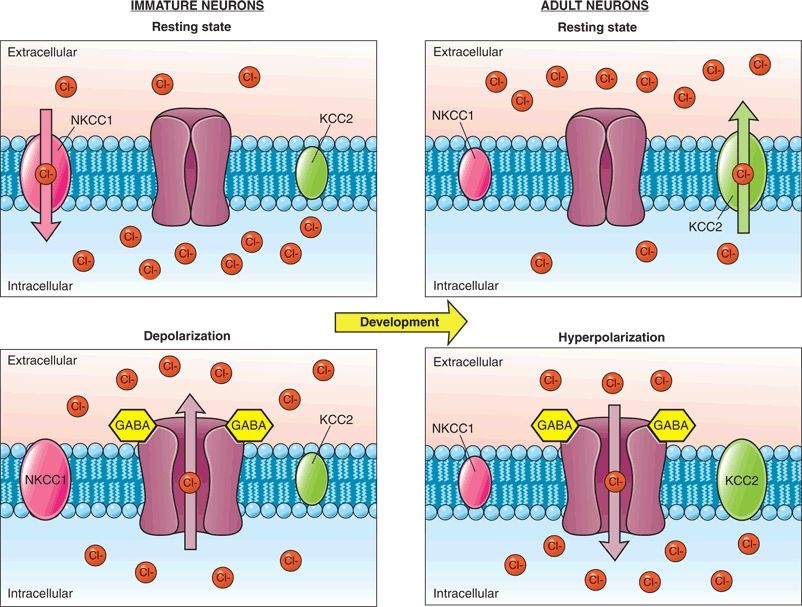
Figure 35.2. Changes in chloride homeostasis during development. In the immature neuron (left portion of panel), the NKCC1 cotransporter is primarily expressed and allows chloride to accumulate in the neuron. When GABA binds to its receptor, the negatively charged chloride ions leave the cell leading to depolarization of the neuron. In the mature neuron (right portion of panel), the chloride cotransporter KCC2 that functions to transport chloride out of the cell predominates. When GABA binds to its receptor, chloride enters the cell causing hyperpolarization of the neuron.
The paradoxical depolarizing effect of GABA during the neonatal period potentially contributes to the refractoriness of neonatal seizures to phenobarbital and benzodiazepines. Interestingly, recurrent neonatal seizures in rodent models lead to an increase in intracellular chloride via a NKCC1-dependent mechanism (16,17). In some experimental models of induced neonatal seizures, inhibition of the NKCC1 transporter with bumetanide alters chloride transport and significantly enhances the anticonvulsant effects of phenobarbital in the neonatal rat hippocampus (17,18). Clinical studies evaluating bumetanide add-on therapy are currently ongoing.
Glutamatergic receptors also regulate excitability in the immature neuron and undergo developmental changes that contribute to the propensity of the neonate to seizures. These receptors can have various functional properties based on their subunit composition that changes during development. N-methyl-d-aspartate (NMDA) receptors in immature neurons express primarily the NR2B subunit that prolongs the duration of the excitatory postsynaptic potential. Increased expression of the NR2C, NR2D, and NR3A subunits confers a reduced sensitivity to blockade by magnesium, resulting in increased excitability (19).
Prognostic Significance
Neonatal seizures are a powerful prognostic indicator of mortality and neurologic morbidity. The summary report from Bergman et al. (2) of 1667 patients noted an overall mortality of 24.7% before 1969 and 18% after 1970. According to Lombroso (20), mortality decreased modestly from about 20% previously to 16% in the early 1980s. These improvements probably reflect better obstetrical management and modern neonatal intensive care. All of these studies relied on seizure diagnosis by clinical criteria and did not require EEG confirmation.
Survivors of neonatal seizures face an exceptionally high risk for cerebral palsy, often with mental retardation and chronic postnatal epilepsy. The National Collaborative Perinatal Population (NCPP) study (21,22) examined numerous clinical perinatal factors for their association with severe mental retardation, cerebral palsy, and microcephaly (Fig. 35.3). The clinical diagnosis of “neonatal seizures” was independently and significantly associated with these adverse outcomes and eclipsed only by “intracranial hemorrhage” in forecasting them.
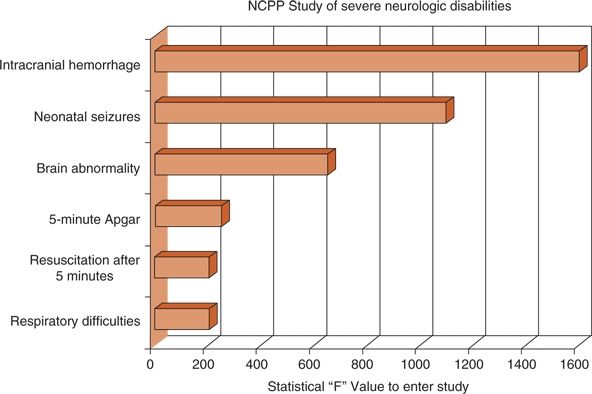
Figure 35.3. The National Collaborative Perinatal Population (NCPP) study prospectively followed more than 34,000 mothers to identify perinatal events associated with adverse outcomes. Fifty neonates were found with subsequent severe neurologic handicaps. Six independent variables, including neonatal seizures, were associated with such neurologically devastating outcomes. (Adapted from Nelson KB, Broman SH. Perinatal risk factors in children with serious motor and mental handicaps. Ann Neurol. 1977;2(5):371–377.)
Contemporary studies of the prognosis after neonatal seizures have emphasized the inclusion of infants whose seizure type was confirmed by EEG monitoring. Pisani et al. (23) identified 106 consecutively admitted neonates with video EEG–confirmed seizures over a 5-year period. The mortality rate was 19% with a favorable outcome found in only 34% of patients. Cerebral palsy was identified in 37% and developmental delay in 34%, and 21% had postnatal epilepsy at 24 months of follow-up.
Of the 207 full-term infants with video EEG–confirmed seizures prospectively enrolled by the Clinical Research Centers for Neonatal Seizures between 1992 and 1997, 28% died (24). Two-year follow-up data were available for 122 patients, or 86% of the survivors. Abnormal neurologic findings were noted in 42%. A Mental Developmental Index (MDI) score below 80 was present in 55%, a Psychomotor Developmental Index (PDI) score <80 in 50%, and chronic postnatal epilepsy in 26%.
Whether seizures themselves adversely affect the developing brain is difficult to determine from clinical studies. Seizure burden may appear to influence outcome because some infants who experience brief, infrequent seizures may have relatively good long-term outcomes, whereas those with prolonged seizures often do not fare as well. However, easily controlled or self-limited seizures may be the result of transient, successfully treated, or benign CNS disorders of neonates, while medically refractory neonatal seizures may stem from more sustained, less treatable, or more severe brain disorders. Legido et al. (25) studied 40 neonates with electrographic seizures detected on randomly timed routine EEG examinations and monitored them for cerebral palsy, mental retardation, and epilepsy. Overall neurologic outcome was more favorable in those with two or fewer seizures per hour than in those with more. In the subgroup with seizures caused by hypoxia–ischemia, cerebral palsy was more frequent when more than five seizures occurred per hour. However, these results might equally reflect more severe underlying injuries that triggered both the additional short-term seizures and greater morbidity on long-term follow-up. In another study, a higher seizure burden was independently associated with a worse neurodevelopmental outcome in hypoxic–ischemic encephalopathy (HIE; see section titled “Diffuse Hypoxic–Ischemic Etiologies” later in this chapter) even after controlling for the severity of hypoxic–ischemic brain injury (26).
A randomized trial was conducted evaluating the utility of treating electrographic seizures detected on amplitude-integrated EEG (aEEG; see section titled “Amplitude-Integrated EEG” later in this chapter). Patients with moderate to severe HIE and clinical seizures were started on aEEG, but they were randomized to treatment of seizures using only clinical evaluation or a combination of aEEG and clinical seizures. There was a trend to shorter seizure duration with treatment of both clinical and electrographic seizures compared to only those observed clinically. In addition, the group also found that a longer duration of seizure was associated with a greater severity of brain injury on MRI (27).
Attempting a balanced approach, McBride et al. (28) followed up 68 high-risk neonates with birth asphyxia, meningitis, and other stressors linked to neonatal seizures. All infants underwent long-term EEG monitoring. Forty developed electrographic seizures, while 28 did not. Based on logistic regression analysis, electrographic neonatal seizures were significantly correlated with death and cerebral palsy. Other investigators (29), using proton magnetic resonance spectroscopy (1H-MRS), found an association of measures of seizure severity with impaired cerebral metabolism measured by lactate/choline and compromised neuronal integrity measured by N-acetylaspartate/choline and suggested this as evidence of brain injury not limited to structural damage detected by magnetic resonance imaging (MRI).
Clinical or EEG-detected seizures are relatively common after newborn heart surgery (NBHS) (30) and generally connote “gray matter” injury or dysfunction. In the Boston Circulatory Arrest Study (BCAS), postoperative EEG seizures were detected in 27/136 (19.8%) and were associated with an 11-point reduction in the Bayley Scales of Infant Development (BSID) at 1-year follow-up (31). By 16 years of age, 139 (87%) of the original cohort of survivors participated in neurocognitive follow-up measuring academic achievement, memory, executive functions, visuospatial skills, attention, and social cognition. After adjustment for all other covariables, the occurrence of seizures in the postoperative period was the medical variable most consistently related to worse outcomes. The scores of both treatment groups tended to be lower than those of the test normative populations, with substantial proportions scoring ≤1 SDs below the expected mean (32).
EEG seizures were also detected commonly after NBHS at Children’s Hospital of Philadelphia. EEG seizures were detected in 11% of 164 infants participating in the APOE polymorphisms study (33). At the 1-year follow-up evaluation, abnormal or suspect examination results occurred in 41/99 (41%) patients without seizures, compared with 11/15 (73%) patients with postoperative seizures (P < 0.027). Moreover, frontal-onset seizures predicted lower MDI scores (P < 0.03), but not lower scores (P < 0.2), compared with non–frontal-onset seizures (34). Neurocognitive testing was repeated in 132 (87%) of 151 survivors at 4 years. The outcomes assessed included cognition, language, attention, impulsivity, executive function, behavior problems, academic achievement, and visual and fine motor skills. Developmental evaluations were performed. After covariate adjustment, occurrence of an EEG seizure was associated with worse executive function (P < 0.037) and impaired social interactions/restricted behavior (P < 0.05). Seizures were not significantly associated with worse performance for cognition, language, attention, impulsivity, academic achievement, or motor skills (all P > 0.1) (35).
Neonatal Seizures Are Harmful to the Neonatal Rodent Brain
Neonatal seizures may be intrinsically harmful to the brain (36). Most seizures were long assumed to be the innocuous, albeit conspicuous, result of an acute injury, and the subsequent long-term neurodevelopmental abnormalities the result of their underlying causes, not the seizures themselves. Basic laboratory studies focused on the effects of seizures on the developing brain have not resolved the controversy (37–41). Immature animals are more resistant than older animals to some seizure-induced injury (42). The immature brain may be resistant to acute seizure-induced cell loss (39); however, functional abnormalities such as impairment of visuospatial memory and reduced seizure threshold (43) occur after seizures, and seizures have been noted to induce changes in brain development, including altered neurogenesis (44), synaptogenesis, synaptic pruning, neuronal migration, and the sequential expression of genes including neurotransmitter receptors and transporters (45,46).
While neonatal seizures seem to induce little histologic damage to the brain (42), studies have revealed that recurrent seizures can produce long-lasting changes in the developing brain, making them more prone to epilepsy and impairing future learning and behavior. Holmes et al. (47) documented impaired spatial learning and memory, decreased activity levels, significantly lower threshold to pentylenetetrazol-induced seizures, and sprouting of CA3 mossy fibers in adult rats that had recurrent neonatal seizures compared to those without neonatal seizures.
Alterations in receptor subunit expression have been implicated as a cause for some of the changes following neonatal seizures. Status epilepticus induced in neonatal rats produced decreased expression of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor GluR2 subunit and increased susceptibility to kainate-induced seizures later in life (48). Another study demonstrated that a single episode of seizures in neonatal rat pups (at P7) produced long-lasting alterations in excitatory glutamatergic synapses that impaired working memory in adulthood (49). Recurrent neonatal seizures likewise produced decreased NR2A expression (50). Hypoxia-induced neonatal seizures appear to decrease long-term potentiation in the hippocampus through an AMPA-mediated mechanism that is suspected to impair synaptic plasticity leading to cognitive impairment (51).
The most frequent clinical setting for the occurrence of neonatal seizures in both term and preterm neonates is following hypoxic–ischemic injury (52). A rodent study of hypoxia-induced seizures demonstrated a decrease in GluR2 receptor expression allowing an increase in calcium influx that may contribute to the chronic epileptogenic effects of hypoxia-induced neonatal seizures (53). Treatment with AMPA receptor antagonists, but not NMDA receptor or GABAA receptor antagonists, after hypoxia-induced seizures in neonatal rats reduced the susceptibility to seizures and seizure-induced injury later in life (54). Likewise, topiramate—which acts mechanistically in part by blocking AMPA/kainate receptors—exerts anticonvulsant activity against perinatal hypoxia-induced seizures (55). These studies highlight the potential utility of topiramate in neonates with seizures associated with hypoxic–ischemic encephalopathy, but the lack of an intravenous formulation makes treatment in critically ill children challenging (56). Nevertheless, in July 2013, the FDA approved the development of an orphan drug formulation of intravenous topiramate, specifically with the neonatal population in mind. Topiramate was found to be safe in one study of infants with HIE who were treated with hypothermia and oral topiramate; however, there was no difference in short-term outcomes (57).
A concern with recurrent neonatal seizures is the concept that “seizures beget seizures”—with recurrent seizures inducing secondary ictal-onset zones. An elegant study by Khalilov et al. (58) attempted to determine if GABA or NMDA signaling was required for creation of a secondary epileptogenic focus. Recurrent seizures were induced in one hippocampus with repeated doses of kainate that eventually propagated to the other hippocampus and established a secondary epileptogenic focus. Addition of an NMDA receptor antagonist to the seizure-naïve hippocampus (not stimulated with kainate) inhibited the creation of a “mirror focus.” Interestingly, GABA-ergic synapses in this system became excitatory in the secondary focus, which is known to be an important contributor to epileptogenesis in the neonatal hippocampus. This study again supports the possible role of NMDA receptor antagonists in the clinical treatment of neonatal seizures.
Finally, neonatal seizures in rats alter the subsequent composition of the GABAA receptor. Each GABAA receptor is a pentamer in which five subunits assemble into a functional ligand-gated receptor (Fig. 35.4). The specific composition of an individual GABAA receptor depends on developmental age. In the studies of Zhang et al. (10), rats with neonatal seizures had a substantially higher proportion of the α1 GABAA subunit than did control animals (Fig. 35.5) (59). Higher levels of the α1 GABAA subunit may provide a protective role in decreasing the severity or frequency of seizures later in life following earlier neonatal seizures (60).
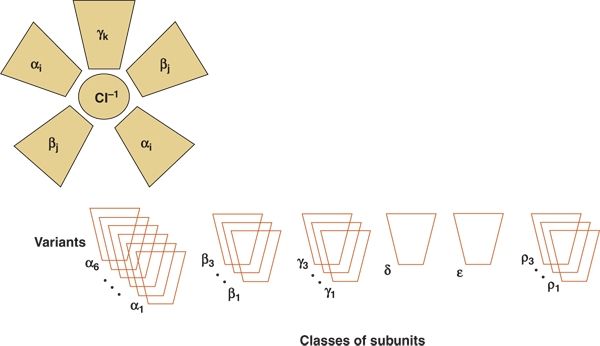
Figure 35.4. The γ-aminobutyric acid receptor is a pentamer structure composed of six possible classes of subunits. The subunits themselves may have multiple variants that are expressed at different developmental ages.
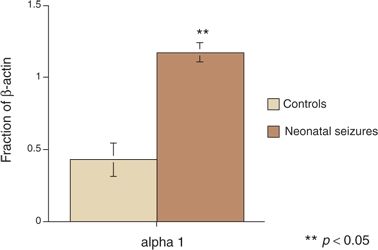
Figure 35.5. Rat pups subjected to seizures had significant differences in α-aminobutyric acid subunit receptor composition in later life compared with control animals. (Adapted from Brooks-Kayal AR, Shumate MD, Jin H, et al. Gamma-Aminobutyric acid (A) receptor subunit expression predicts functional changes in hippocampal dentate granule cells during postnatal development. J Neurochem. 2001;77(5):1266–1278.)
CLASSIFICATION AND CLINICAL FEATURES OF NEONATAL SEIZURES
Application of a syndromic classification to neonatal seizures is limited when considered in light of the classification of the International League Against Epilepsy (ILAE) (61). Almost all neonatal seizures are thought to be symptomatic, an acute reaction, or consequence of a specific etiology. The most recent ILAE classification addresses only five neonatal syndromes: benign neonatal seizures, benign familial neonatal epilepsy (BFNE), early myoclonic encephalopathy (EME), Ohtahara syndrome, and migrating partial seizures of infancy (61). These are discussed later.
Seizures in the neonate are uniquely different from those in older infants and children. These differences are based on mechanisms of epileptogenesis, the developmental state of the immature brain, and the relatively greater importance of nonepileptic mechanisms of seizure generation in this age group. Neonatal seizures may be classified by (i) clinical manifestations, (ii) the relationship between clinical seizures and electrical activity on the electroencephalogram, and (iii) seizure pathophysiology.
Clinical Classifications
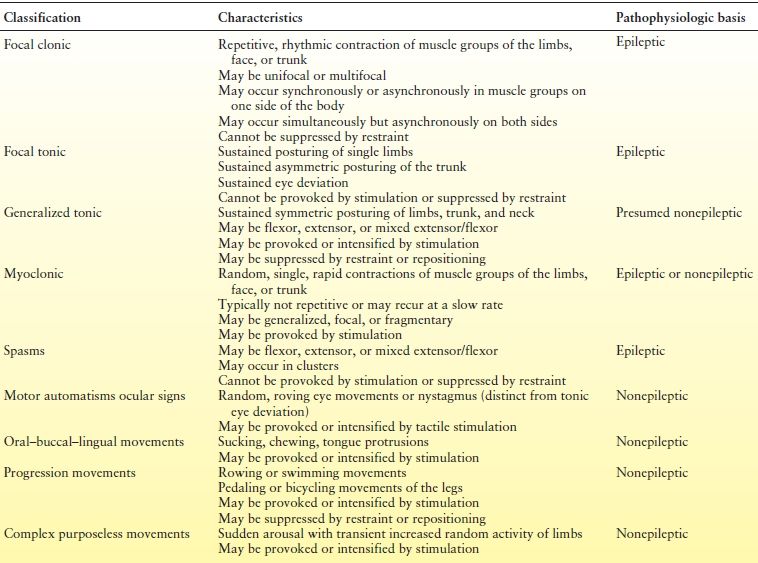
A number of clinical classifications of neonatal seizures have been published (62–66). Early classifications focused on the clinical differences between seizures in neonates and those in older children: neonatal seizures were reported to be clonic or tonic, not tonic–clonic; when focal, they were either unifocal or multifocal. Later classifications included the term myoclonus. Another distinguishing feature of neonatal seizures is the occurrence of events described initially as “anarchic” (62) and thereafter “minimal” (67) or “subtle” (65). These events included oral–buccal–lingual movements such as sucking and chewing; movements of progression, such as bicycling of the legs and swimming movements of the arms; and random eye movements. First considered epileptic in origin, they were later deemed to be exaggerated reflex behaviors and thus were called “brainstem release phenomena” or “motor automatisms” (64). Table 35.1 lists the clinical characteristics of neonatal seizures according to a current classification scheme (68) that can be applied through observation.
Table 35.1 Clinical Characteristics, Classification, and Presumed Pathophysiology of Neonatal Seizures
Seizure Pathophysiology
Clinical seizures may be classified as epileptic or nonepileptic (Table 35.2). Some clinical neonatal seizures are clearly epileptic, occurring in close association with EEG seizure activity, involving clinical events that can be neither provoked by stimulation nor suppressed by restraint and directly triggered by hypersynchronous cortical neuronal discharges. The following properties of the developing brain intensify seizure initiation, maintenance, and propagation: increases in cellular and synaptic excitation and a tendency to enhance propagation of an epileptic discharge (40,69–71). The clinical events that are most clearly epileptic in origin are focal clonic, focal tonic, some types of myoclonic, and rarely spasms (see Tables 35.1 and Table 35.2). Electrical-only seizures (also called subclinical or occult) are, by definition, epileptic.
Table 35.2 Classification of Neonatal Seizures by Electroclinical Findings

Best considered nonepileptic in origin (64) are events that occur in the absence of electrical seizure activity but that have clinical characteristics resembling reflex behaviors. Such clinical events, whether provoked by stimulation or arising spontaneously, can be suppressed or altered by restraining or repositioning the infant. The clinical events may grow in intensity with increases in the repetition rate of stimulation (temporal summation) or the sites of simultaneous stimulation (spatial summation). Some types of myoclonic events, generalized tonic posturing, and motor automatisms can be classified as “nonepileptic” (see Tables 35.1 and 35.2).
Paroxysmal clinical changes related to the autonomic nervous system have been proposed as manifestations of some seizures. These include stereotyped, episodic alterations in heart rate, respiration, and blood pressure (72–74). Skin flushing, salivation, apnea (75,76), and pupillary dilation may also be autonomic signs of seizures, but they are usually associated with other clinical manifestations, except in the therapeutically paralyzed infant (64).
Electrographic Seizures
Although visual observation is critical to the detection of clinical neonatal seizures, the electroencephalogram offers the most important means of confirmation and characterization. Infants with normal background activity are much less likely to develop seizures than are those with significant background abnormalities (77).
Interictal Background and Prediction Value
The ongoing cerebral electrical activity is the stage on which the drama of the episodic electrographic seizure unfolds. In many ways, the integrity of the EEG background is as critical than the mere presence or absence of the seizures themselves. For example, with or without electrographic seizures, an extremely abnormal EEG background (burst suppression or isoelectric recording) inherently conveys a sense of profound electrophysiologic disruption and forecasts a high risk for death or adverse neurologic outcome. Conversely, a nearly normal interictal EEG background suggests relatively preserved neurologic health despite the unwanted intrusion of the seizures.
The interictal background also occasionally can offer clues to seizure etiology. Persistently unifocal sharp waves (arising from a very limited region of the cortex) may suggest a restricted injury such as localized subarachnoid hemorrhage, contusion, or stroke, whereas multifocal sharp waves suggest diffuse dysfunction. Hypocalcemia is a consideration if a well-maintained background features excessive bilateral central spikes. Inborn errors of metabolism, such as maple syrup urine disease, are sometimes associated with distinctive vertex wicket spikes. Pseudoperiodic discharges raise the suspicion of herpes simplex virus encephalitis or an acute destructive localized lesion such as stroke or hemorrhage. A grossly abnormal electroencephalogram in the absence of any obviously acquired disease suggests cerebral dysgenesis.
Interictal EEG spikes per se have uncertain diagnostic significance (78). Interictal focal sharp waves and spikes are not typically considered indicators of epileptogenesis in the same way as they are in older children and adults. Compared with those of age-matched neonates without seizures (79,80), the interictal records of infants with electroencephalogram-confirmed seizures have background abnormalities, excessive numbers of “spikes” (lasting <200 msec) compared with sharp waves (lasting >200 msec), excessive occurrence of spikes or sharp waves per minute, and a tendency for “runs,” “bursts,” or “trains” of repetitive sharp waves. However, only a few infants with confirmed seizures exhibit all of these interictal characteristics, and many show no excessive spikes or sharp waves. Routine duration interictal EEGs are inadequate to characterize neonatal seizure burden (81).
Characteristics
At the heart of the epileptic process is the abnormal, excessive, repetitive electrical firing of neurons. Affected neurons lose their autonomy and are engulfed by the synchronized bursts of repeated electrical discharges. As excitatory neurotransmission fails, the electrophysiologic cascade is interrupted and the seizure ends. Electrographic seizures in the neonate have varied appearances and are relatively rare before 34 to 35 weeks conceptional age. The morphology, spatial distribution, and temporal behavior of the seizure discharges may differ within and between individuals. Despite the differences between the term and preterm neonatal brain, the variety of types of electrographic seizures does not differ between them (82).
Morphology
An electrographic seizure is a discrete abnormal event lasting at least 10 seconds, with a definite beginning, middle, and end (83). No single morphologic pattern characterizes a seizure (Fig. 35.6). Even in the same patient, the ictal EEG activity may appear pleomorphic. The “typical” neonatal seizure begins as low-amplitude, rhythmic, or sinusoidal waveforms or spike or sharp waves. As the seizure evolves, the amplitude of the ictal activity increases, while its frequency slows (84). Spikes or sharp waves are not necessarily present. Instead, rhythmic activity of any frequency (delta, theta, alpha, or beta) can make up the ictal patterns at the scalp surface.
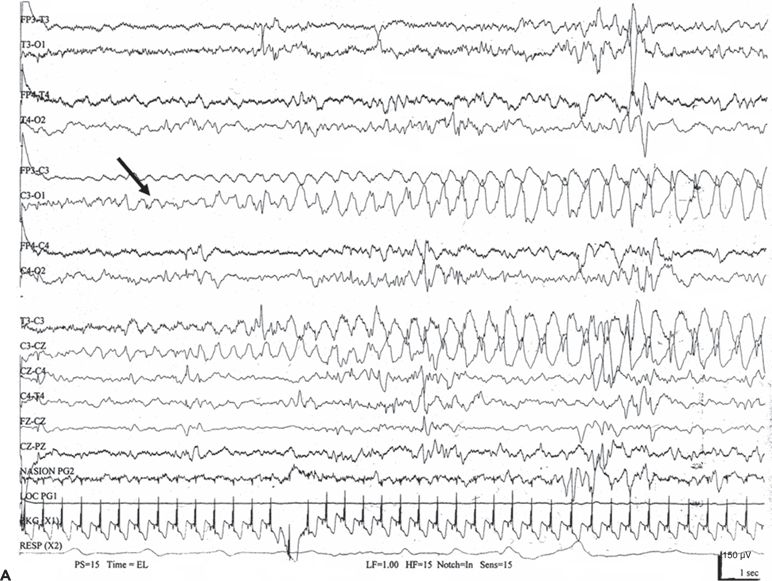
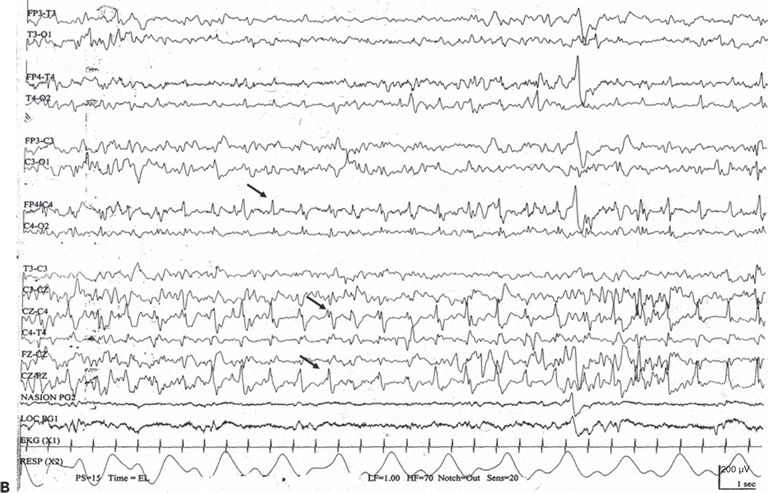
Figure 35.6. No single morphologic pattern characterizes electrographic neonatal seizures; rather, their distinctive behavior as a discrete, evolving electrographic event identifies them as ictal. A: A focal seizure arises from C3 (arrow) as low-amplitude, rhythmic theta activity that gradually changes to higher- amplitude delta activity. B: An electrographic seizure is in progress as repetitive spikes in the right frontopolar, central, and midline vertex regions (arrows).
Spatial Distribution
In older children, generalized seizures may appear simultaneously, synchronously, and symmetrically in both hemispheres. In the neonatal brain, which lacks the physiologic organization necessary for such exquisite orchestration, individual seizures always arise focally and then migrate and spread. Seizures may also migrate from one hemisphere to another (68). Occasionally, simultaneous focal seizures may appear to behave independently, spreading to all brain regions and superficially masquerading as a “generalized seizure.” However, the ictal patterns are not those of the truly generalized seizures.
Diffuse causes of encephalopathy such as meningitis, hypoglycemia, or hypoxia–ischemia may cause focal seizures or induce multiple seizures that each originate from different scalp regions—so-called multifocal-onset seizures; those that arise from the same scalp location are unifocal onset and raise the possibility of a localized structural abnormality such as a stroke (83), reflecting the restricted functional disturbance.
Temporal Profile
The typical duration of an individual electrographic neonatal seizure is about 1 to 2 minutes and is followed by an interictal period of variable length (Fig. 35.7). These temporal characteristics were obtained from relatively brief tracings randomly selected during a variety of acute encephalopathies (8). A study of the temporal characteristics of 15 patients with HIE found the median seizure duration to be 206 seconds (interquartile range IQR 98 to 331 seconds) (85). Eleven of 15 patients had a total seizure duration in excess of 30 minutes per hour, a commonly used definition for neonatal status epilepticus, since solitary, prolonged electrographic seizures are uncommon in newborns. Repetitive brief serial seizures are much more characteristic than prolonged seizures lasting many hours. Other investigators have also reported status epilepticus during therapeutic hypothermia for neonatal HIE (86).
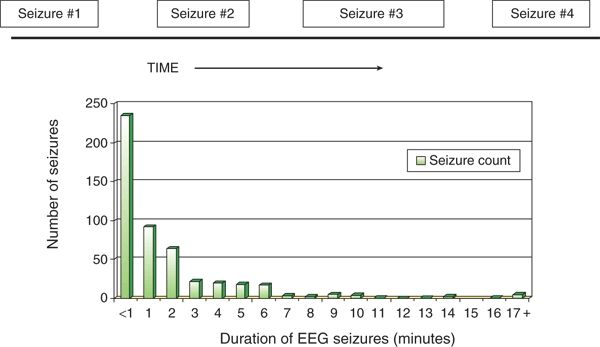
Figure 35.7. In most neonates with electrographic seizures, the electroencephalogram shows a series of brief ictal events, typically lasting <2 minutes, followed by varying- length interictal periods. The histogram shows the distribution of durations (minutes) of 487 electroencephalographic seizures recorded from 42 neonates. (Adapted from Clancy RR, Legido A. The exact ictal and interictal duration of electroencephalographic neonatal seizures. Epilepsia. 1987;28(5):537–541.)
Neonates may demonstrate events that have morphologic characteristics of electrographic seizures with a duration of <10 seconds. These discharges have been termed brief rhythmic discharges (BRDs) or brief intermittent rhythmic discharges (BIRDs) (87). The significance of these discharges is unclear. A study of 340 subjects noted that BIRDs had a higher association with leukomalacia or hypoglycemia and were associated a higher risk of neurodevelopmental abnormalities (88). Another limited study found no significant difference in mortality and neurodevelopmental outcome for subjects with BIRDs or electrographic seizures; however, subjects with BIRDs only were less likely to have postnatal seizures (89). Given their frequent association with clear electrographic seizures, care should be taken to scrutinize the EEG closely for seizures.
Special Ictal Electroencephalographic Morphologies
Some ictal patterns unique to the neonatal period are associated with severe encephalopathies. Electrical seizures of the depressed brain are long, low in voltage, and highly localized. They may be unifocal or multifocal and show little tendency to spread or modulate. Not associated with clinical seizures, they occur when the EEG background is depressed and undifferentiated and suggest a poor prognosis. Alpha (α) seizure activity (90–92) is characterized by sudden, transient, rhythmic activity in the α frequency range (8 to 12 Hz) in the temporal or central region, unaccompanied by clinical events. An α discharge usually indicates a severe encephalopathy and poor prognosis.
Video-EEG monitoring has been the basis of clinical investigations into the classification, therapy, and prognosis of neonatal seizures (93–95). It has become more widely available, but remains limited at many locations.
Amplitude-Integrated EEG
Amplitude-integrated EEG (aEEG) is becoming increasingly used in the neonatal intensive care unit setting for bedside evaluation of cerebral activity. While various techniques are available, limited arrays of 2 or 4 two electrodes are commonly used to acquire data that are then processed and compressed to provide a simple trend of the background EEG activity in those regions (Fig. 35.8). Advantages over conventional EEG include widespread availability, ease of application, and the lack of dependence on specially trained neurophysiologists. The compressed data provides information about the background of the EEG that can be used for prognostic purposes, including inclusion in therapeutic neuroprotection (e.g., cerebral head cooling) (96). There is acceptable agreement between conventional EEG and aEEG background classifications when the tracings are normal or when markedly abnormal. For example, both techniques would correctly identify a flat or burst suppression tracing. However, the agreement between these two techniques is much poorer among the middle-ground, “moderately abnormal” aEEGs (97).
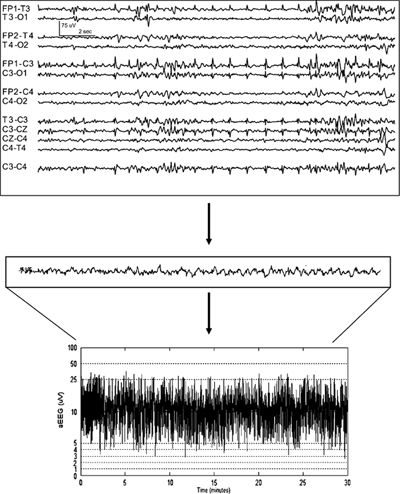
Figure 35.8. The routine neonatal EEG examination typically displays 12 or more channels from the full array of the 10 to 20 system. Cerebral function monitors, such as aEEG, use a single channel from a pair of scalp electrodes (commonly the left and right parietal regions) and then process the raw EEG to a compressed display, which is very useful for reviewing long-term trends.
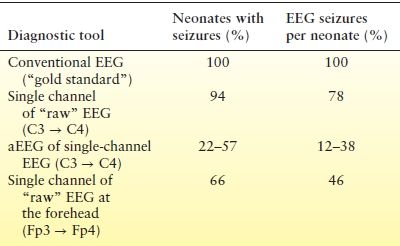
Neonatal seizures can be detected with aEEG as sudden elevations of the lower and upper margins of the background tracing (Fig. 35.9). Studies comparing neonatal seizure detection between aEEG and conventional EEG demonstrate that only 22% to 57% of infants with seizures are accurately identified by neonatologist readers. These readers correctly detected 12% to 38% of the seizures confirmed by conventional EEG (Table 35.3) (98,99). Certain seizure characteristics influence their identification: amplitude, duration, number of seizures per hour, and the spatial restrictions imparted by sampling just 1 or 2 electrode pairs (99). Clearly, aEEG cannot supplant conventional EEG but can provide very useful complementary information that may guide decision making at the bedside in real time.
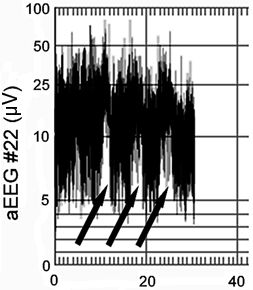
Figure 35.9. Thirty-minute aEEG in a term infant with three captured seizures (arrows).
Table 35.3 Comparisons of Conventional and Amplitude-Integrated EEG for Seizure Detection
Continuous EEG Monitoring (cEEG) of Neonates
The ACNS has published guidelines that provide a recommendation for monitoring neonates in the ICU (100). Listed indications for cEEG include evaluation for electrographic seizures and to judge the severity of encephalopathy. The committee recommended monitoring of high-risk neonates for seizures for 24 hours and, if seizures are detected, until the patient is seizure free for at least 24 hours. The ACNS has also published a guideline for standardized terminology and categorization of cEEG in neonates (101).
Measures of Electrographic Seizure “Burden”
Most electrographic neonatal seizures do not provoke distinctive clinical signs (102) (Fig. 35.10). It would be useful to develop measures of the “burden” of electrographic seizures in individual infants.
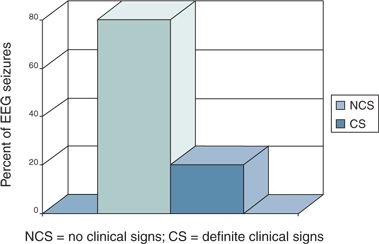
Figure 35.10. In one study, only 20% of electrographic neonatal seizures produce definite clinical signs. (Adapted from Clancy RR, Legido A, Lewis D. Occult neonatal seizures. Epilepsia. 1988;29(3):256–261.)
The simplest measure is to simply consider them dichotomously “present” or “absent.” However, the number of recorded electrographic seizures varies widely, so a simple seizure frequency count could be used (Fig. 35.11). For example “there were four seizures in the 1st hour and two in the 2nd hour of monitoring.” Because individual electrographic seizures also vary in length, another measure of EEG seizure burden is to describe the percentage of time in which seizure activity is present in any brain region. This can range from a 0% if no seizures are captured to 100% if the entire record demonstrates seizure activity anywhere in the brain. Unfortunately, there is only a modest (albeit statistically significant) correlation between seizure frequency counts and the percentages of the recordings showing seizure activity (99). The most detailed measure of seizure burden further incorporates knowledge of their numbers, durations, and spatial distributions. Individual electrographic seizures may remain confined to their area of origin or may spread substantially to other regions (103). This varies considerably among individual neonates (Fig. 35.12) (95). A comprehensive approach to measure EEG seizure burden, temporal–spatial analysis, reduces the entire neonatal montage into five nonoverlapping areas of interest (104): (Fig. 35.13). The percentage of ictal time at each of these five regions gives the most comprehensive picture of the geographic distribution of seizure burden. Future investigations may determine whether a “dose–response” curve exists between this fuller, temporal–spatial measure of seizure burden and eventual long-term neurodevelopmental follow-up.
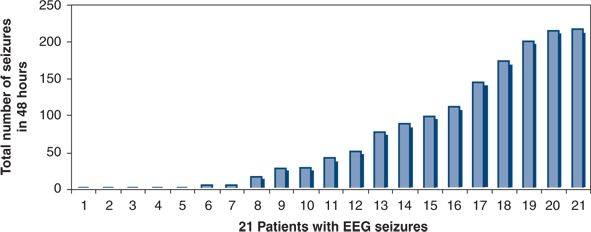
Figure 35.11. Distribution of the total number of electrographic neonatal seizures during 48 hours of electroencephalograph monitoring after newborn heart surgery. From 1 to 217 seizures occurred during the study period. (Adapted from Sharif U, Ichord R, Saymor JW, et al. Electrographic neonatal seizures after newborn heart surgery. Epilepsia. 2003;44:164.)
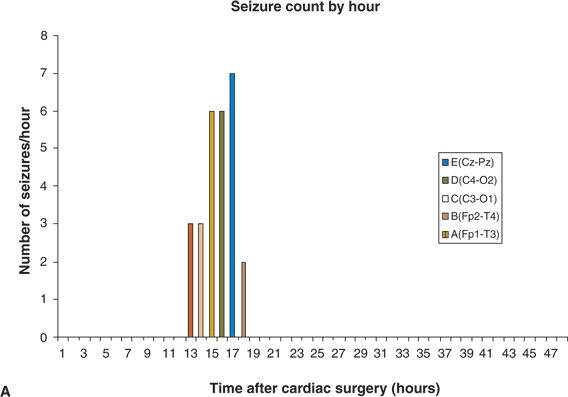
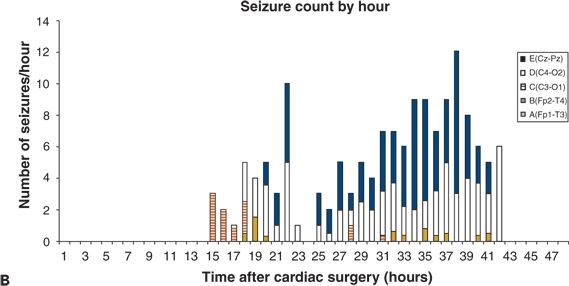
Figure 35.12. The spatial distribution of electroencephalographic (EEG) seizures varies among neonates. A: All EEG seizures begin in a single brain region (C4–O2). B: EEG seizures begin in four locations (Cz–Pz, C4–O2, C3–O1, and Fp2–T4). (Adapted from Sharif U, Ichord R, Saymor JW, et al. Electrographic neonatal seizures after newborn heart surgery. Epilepsia. 2003;44:164.)
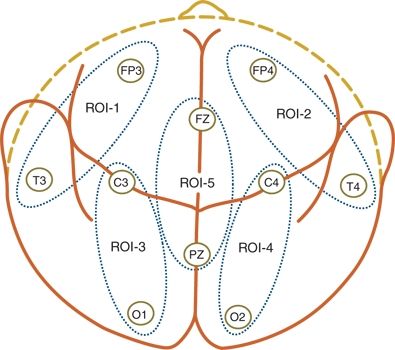
Figure 35.13. The entire array of the standard neonatal electroencephalogram can be reduced to five nonoverlapping regions of interest that identify the spatial characteristics of electroencephalographic seizures.
ETIOLOGIC FACTORS
Acute or chronic conditions can give rise to seizures. In most cases, specific causes can be determined after analysis of clinical and laboratory information (Table 35.4). Table 35.5 lists potential causes of neonatal seizures, but only a few are discussed in detail.
Table 35.4 Data to Determine the Etiology of Neonatal Seizures
aVaries by US states.
bIn the proper clinical context.
Table 35.5 Etiologies of Neonatal Seizures

Acute Causes
Diffuse Hypoxic–Ischemic Etiologies
Probably the most common cause of neonatal seizures, “acute neonatal encephalopathy” (105) is characterized by depressed mental status (lethargy or coma), seizures, axial and appendicular hypotonia with an overall reduction in spontaneous motor activity, and clear evidence of bulbar dysfunction with poor sucking, and swallowing, and an inexpressive face (106). Care should be taken to separate this generic designation from neonatal HIE. Not every infant who is acutely encephalopathic has suffered hypoxia–ischemia (107). The American College of Obstetricians and Gynecologists Task Force on Neonatal Encephalopathy and Cerebral Palsy suggests four diagnostic criteria for HIE: (i) evidence of metabolic acidosis in fetal umbilical cord arterial blood obtained at delivery (pH value <7 and base deficit >12 mmol/L), (ii) early onset of severe or moderate neonatal encephalopathy in infants born at 34 or more weeks of gestation, (iii) subsequent cerebral palsy of the spastic quadriplegic or dyskinetic type, and (iv) exclusion of other identifiable etiologies such as trauma, coagulation disorders, infectious conditions, or genetic disorders (108). These four conditions should occur in the context of a “sentinel” hypoxic event immediately before or during labor, such as uterine rupture, abruption of the placenta, or prolapse of the umbilical cord. There should also be a sudden and sustained fetal bradycardia or the absence of fetal heart rate variability; persistent, late, or variable decelerations; Apgar scores of 0 to 3 after 5 minutes; and, in most, multisystem involvement within 72 hours of birth. Examples of multisystem malfunction (109) include acute renal tubular necrosis, elevated values of liver function tests, necrotizing enterocolitis from bowel ischemia, and depressed blood cell lines (e.g., thrombocytopenia) because of ischemic injury of the bone marrow (110). Early imaging studies should show acute diffuse cerebral abnormalities consistent with hypoxia–ischemia. A recent intriguing investigation in this specific hypoxic–ischemic scenario is the development of intraneuronal alkalosis as a specific trigger for seizures, which can be treated by unique paradigms of ventilatory management in which normocapnia was gradually, rather than abruptly, restored (111,112).
Other conditions that can clinically mimic acute neonatal HIE are some of the inborn errors of metabolism, pyridoxine dependency, stroke, coagulopathies, sinovenous thrombosis, and “fetal sepsis syndrome” (113–116), which can occur with sepsis or chorioamnionitis (117,118). The latter is suspected in a mother with abdominal pain and tenderness, fever, leukocytosis, and foul-smelling amniotic fluid and can be confirmed by pathologic examination of the placenta and umbilical cord (119).
Perinatal stroke is defined as a cerebrovascular event occurring between 28 weeks of gestation and 7 days of age. The incidence is 1 in 4000 live births (120). There are two main clinical presentations: (i) acute appearance of neonatal seizures, hypotonia, feeding difficulties, and, rarely, hemiparesis (83,119,121,122) (Fig. 35.14) and (ii) later discovery of stroke through the gradual appreciation of a congenital hemiparesis or the onset of a partial seizure disorder in an infant apparently healthy at birth. Risk factors include congenital heart defects (CHDs), blood and lipid disorders, infection, placental disorders, vasculopathy, trauma, dehydration, and extracorporeal membrane oxygenation (ECMO).
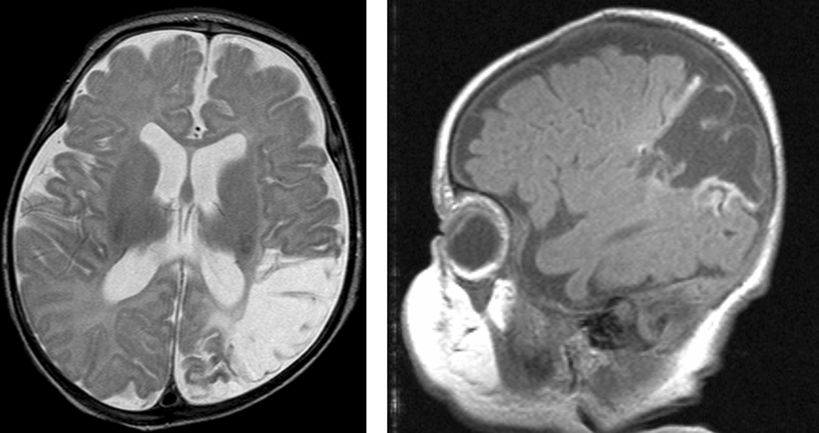
Figure 35.14. Arterial ischemic stroke in the distribution of the left middle cerebral artery in a 41-week–estimated gestational age infant with a prothrombotic disorder.
Cerebral sinovenous thrombosis is estimated to occur at a rate of 0.67 cases per 100,000 children per year (123,124) in neonates and older children. The neonatal presentation most frequently includes seizures (57% to 71%) and other nonspecific CNS signs such as lethargy (35% to 58%), but only infrequently frank hemiparesis (123,125) (Fig. 35.15). Maternal risk factors associated with thrombosis included preeclampsia/hypertension, gestational diabetes, and meconium aspiration or meconium-stained placenta (125). The sagittal and transverse sinuses are most commonly involved, but multiple sinus thromboses also occur. The reported outcomes include a variable mortality rates from 2% to 13%; 21% developed normally while 60% had cognitive impairment, 64% had motor impairment, and 40% had epilepsy (125,126).
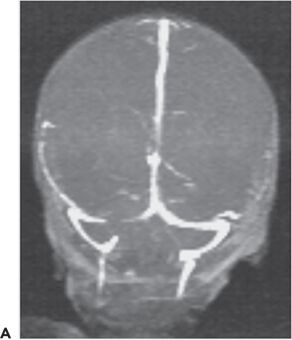
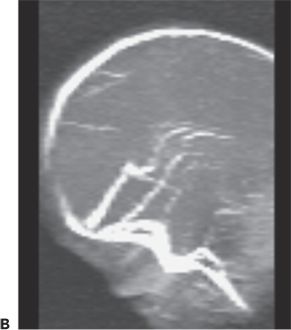
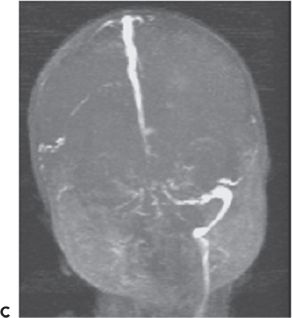
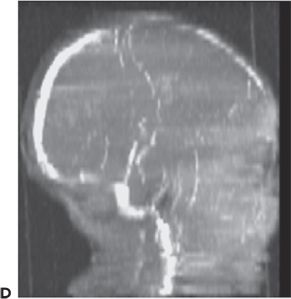
Figure 35.15. Magnetic resonance venogram of a 2-week-old term infant admitted for seizures, lethargy, and dehydration. A,B: Thrombosis of the right transverse sinus was noted on the 1st day of hospitalization. C,D:By day 10, the thromboses had extended to the sigmoid, jugular, and straight sinuses.
ECMO is an effective therapy for newborn infants with life-threatening respiratory failure unresponsive to maximum conventional medical support. However, the procedure requires ligations of the right common carotid artery and right jugular vein at a time when the infants’ underlying lung disease may render them particularly vulnerable to the effects of diffuse CNS hypoxia–ischemia. The high rate of subsequent neurologic morbidity among survivors raises the possibility that ECMO itself may contribute to ischemic–reperfusion brain injuries (127). A high proportion of survivors have MRI-identified focal parenchymal brain lesions, often announced by seizures during ECMO. Cerebral hemorrhage and infarction have been reported in 28% to 52% of ECMO-treated infants (128).
CHDs enhance the risk for neonatal seizures (129), which can arise preoperatively or postoperatively. Strokes may occur from multiple mechanisms including right-to-left intracardiac shunting or embolization during cardiac catheterization (130). CHDs also may be associated with the presence of other midline somatic defects including CNS anomalies. Seizures can arise from concurrent cerebral dysgenesis as well (131). Hypocalcemia may trigger seizures in the setting of DiGeorge syndrome. However, seizures usually arise after NBHS; they do not occur at random, but, rather, are influenced by suspected or confirmed genetic disorders, aortic arch obstruction, or the need for prolonged deep hypothermic circulatory arrest (132). This population is especially valuable for neuroprotection trials, because the child’s status can be determined before surgery (133).
Metabolic Etiologies
Hypoglycemia, hyponatremia, hypernatremia, hypocalcemia, hypomagnesemia, and acute hyperbilirubinemia (acute kernicterus) can be associated with neonatal seizures. These conditions are detectable by simple screening tests (see Table 35.4).
Hypoglycemia may itself cause brain damage independent of the seizures. Causes of hypoglycemia that should be evaluated in children include simple prematurity, maternal diabetes, nesidioblastosis, galactosemia, defects of gluconeogenesis, glycogen storage diseases, and respiratory chain defects. Glucose transporter type I syndrome (GLUT I deficiency) is characterized by infantile seizures that usually begin between 6 and 12 weeks of life, developmental delay, ataxia, and progressive microcephaly (134–136). Affected newborns appear normal at birth. Neonatal seizures, initially rare, increase in frequency as the developmental delay becomes evident. The resultant diminution in transported glucose at the blood–brain barrier markedly reduces brain and cerebrospinal fluid values. Genetic studies often implicate mutations in SLC2A1. The ketogenic diet is a highly effective treatment modality since it provides an alternate form of fuel for the CNS.
Inborn Errors of Metabolism
For a detailed discussion, see Chapter 31. The discussion below is limited to the diagnosis of common neonatal conditions amenable to treatment with a specific intervention.
Maple syrup urine disease, ketotic and nonketotic hyperglycinemia, and urea cycle disorders may all induce a severe acute encephalopathy with seizures. Nonketotic hyperglycinemia has a catastrophic clinical presentation (aptly named glycine encephalopathy) with intractable seizures, coma, hiccups, apnea, pupil-sparing ophthalmoparesis, spontaneous and stimulus-provoked myoclonus, and a burst suppression pattern on electroencephalography (137). Glycine levels are elevated in the blood and cerebrospinal fluid. The disorder represents an inability to cleave glycine, which is both an excitatory and inhibitory neurotransmitter. Treatment involves an NMDA receptor antagonist, as well as magnesium, sodium benzoate, and dextromethorphan.
The ketotic hyperglycinemias and propionic and methylmalonic acidemias present with overwhelming multisystem failure and dehydration, ketoacidosis, and fulminant CNS signs such as seizures, vomiting, and coma. Diagnosis is made by serum amino acid surveys and measurement of specific enzyme activity.
Carbamoyl phosphate synthetase deficiency, ornithine carbamoyltransferase deficiency, citrullinemia, and argininosuccinic acidemia are among the large number of urea cycle abnormalities, and each causes neonatal seizures in the 1st days or weeks of life. Coma and prominent bulbar dysfunction are noted with ophthalmoparesis, fixed pupils, absent gag reflex, poor sucking, and apnea. The degree of serum ammonia elevation may correlate with the discontinuity in the abnormal EEG backgrounds (138).
Biotinidase deficiency may produce alopecia, seborrheic dermatitis, developmental delay, hypotonia, and ataxia. Seizures may begin as early as the 1st week of life. The diagnosis is made by measurement of blood levels of biotinidase activity. Oral administration of free biotin daily is the treatment.
Pyridoxine-dependent seizures (139,140) usually arise between birth and 3 months of age, although atypical cases have been reported up to 3 years. Some seizures can be appreciated in utero (141). The neonate presents with agitation, irritability, jitteriness, diminished sleep, and intractable clonic seizures. The EEG patterns are entirely nonspecific and include abnormal backgrounds, excessive multifocal sharp waves, and focal electrographic seizures evolving to hypsarhythmia later in the 1st year. Intravenous administration of 50 to 100 mg of pyridoxine causes cessation of seizures and disappearance of epileptiform EEG activity within a few hours of administration. Lifelong therapy with pyridoxine 50 to 100 mg/day is necessary. Despite early treatment, some neonates are eventually retarded and show MRI evidence of a leukodystrophy. Mutations in α-aminoadipic semialdehyde (α-AASA) dehydrogenase (antiquitin) leading to inactivation of pyridoxal phosphate (PLP) have been found in multiple individuals with pyridoxine-dependent seizures (142). PLP is an essential cofactor in multiple enzymatic reactions, including the formation of GABA. Elevations of urinary α-AASA can be used as a screening tool for identifying individuals with antiquitin mutations. However, this should not substitute for a pyridoxine trial, especially in the acute setting. Mutations in pyridox(am)ine 5′-phosphate oxidase may cause early neonatal seizures due to decreased synthesis of PLP. Treatment with PLP is necessary to overcome this defect (143).
Folinic acid–responsive neonatal seizures were first described by Hyland as the unexpected appearance of seizures in term infants during the first few hours or days of life (144). Subsequently intractable, the seizures were associated with severe developmental delay, progressive atrophy on MRI examination, and frequent bouts of status epilepticus. Analysis of cerebrospinal fluid by means of high-performance liquid chromatography with electrochemical detection consistently revealed a characteristic peak that aided in diagnosis. Seizures ceased and the EEG pattern improved after the administration of 2.5 mg of folinic acid twice daily. Gallagher et al. (145) identified the biochemical marker for folinic acid–responsive seizures in two individuals who were controlled with pyridoxine. They identified gene mutations in antiquitin in those two individuals along with seven other individuals with folinic acid–responsive seizures. The authors suggest that the two conditions are allelic and recommend considering treating patients with α-AASA dehydrogenase deficiency with both pyridoxine and folinic acid.
Deficiency of the molybdenum cofactor and isolated sulfite oxidase deficiency are autosomal recessive errors that produce severe neurologic symptoms resulting from a lack of sulfite oxidase activity (146). The presentation includes poor feeding, an abnormally pitched cry, jitteriness, and intractable seizures with the presence of sulfites in the urine. Synthesis of molybdenum cofactor requires the activities of at least six gene products including gephyrin (147), a scaffolding protein present at inhibitory synapses. Unfortunately, there is no effective treatment, and prognosis for neurologic recovery and survival is poor.
Neonatal Intoxications
Lidocaine or mepivacaine inadvertently injected into the fetal scalp during local pudendal analgesia for the mother, cocaine, heroin (148), amphetamines, propoxyphene, and theophylline also may cause seizures.
Chronic Causes
Cerebral Dysgenesis
Some neonatal seizures result from long-standing disorders, such as cerebral dysgenesis, neurocutaneous syndromes, genetic disorders, or very early onset epilepsy. An MRI scan should be performed early to uncover cerebral dysgenesis (149). In lissencephaly or hemimegalencephaly (Fig. 35.16), no acute cause for seizures such as neonatal depression or birth trauma is present, and the infant appears outwardly well yet experiences seizures. The identification of cerebral dysgenesis on neuroimaging should not dissuade the clinician from seeking evidence of inborn errors of metabolism, as both may coexist (e.g., cytochrome oxidase deficiency, glutaric aciduria types I and II, 3-hydroxyisobutyric aciduria, 3-methylglutaconic aciduria, 3-ketothiolase deficiency, sulfite oxidase deficiency, pyruvate dehydrogenase deficiency, neonatal adrenoleukodystrophy, fumaric aciduria, long ketotic hyperglycinemia, and Zellweger syndrome) (150).
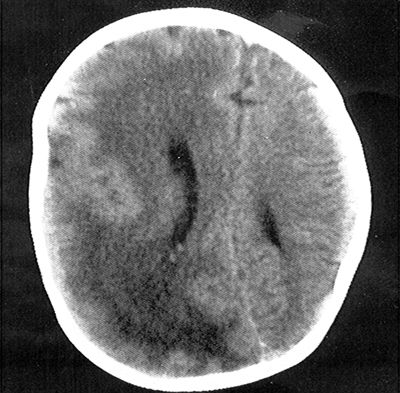
Figure 35.16. Computed tomography scan of the head showing right hemimegalencephaly with dysplastic and enlarged right cerebral hemisphere. Brain magnetic resonance imaging provides better resolution and definition of the abnormality and reveals subtle involvement of the contralateral hemisphere.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


