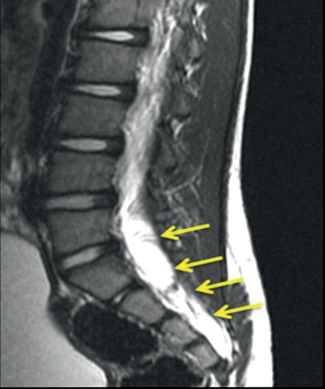
73 What is myelomeningocele? Extrusion of CNS structures (neural tissue, CSF, and meninges) through a posterior spinal defect without any skin covering it1,2 What other terms are used synonymously with myelomeningocele? Open spinal dysraphism, spina bifida aperta, spina bifida cystica1,2 What is the general pathophysiology of the formation of myelomeningocele? Incomplete closure of caudal neural tube from heterogeneous etiologies that cause failure of pertinent molecular expression on neuroecto dermal cell surface2 What are some possible etiologies responsible for myelomeningocele? • Splotch (Sp) mutation in Pax-3 gene (gene expression regulator), which is expressed in neural folds during neural tube closure • Maternal folate deficiency • Teratogens What is the disrupted neural tissue at the site of a neural tube defect called? Neural placode2 How does the placode correspond to the location of the myelomeningocele? • Segmental placode is lumbar, thoracolumbar, and thoracic • Terminal placode is lumbosacral and sacral How do cervicothoracic myelomeningoceles differ from lumbosacral myelomeningoceles? Cervicothoracic myelomeningoceles • are covered by skin and/or tough fibrous tissue and are thus closed lesions and do not leak CSF. • have little to no fascial and posterior bony defects. • have a small sliver of nerve tissue running through the fascial defect from the dorsum of the spinal cord to the inner surface of the sac. • cause little to no motor and sensory deficits.1 What is the incidence of myelomeningocele? 0.7–0.8/1000 live births, with regional variance (Ireland: 5/1000 live births, versus native Africans: 0.1/1000 live births)1 What factors have contributed to the drop in myelomeningocele incidence in the U.S. (from 0.6/1000 live births to 0.3/1000 live births)? • Better maternal nutrition (folate supplementation from prenatal multivitamins) • Easier access to prenatal diagnosis with simultaneous increase in elective terminations • Unknown factors1 Is there a gender preference? Yes, 1:3 male/female1 How is myelomeningocele diagnosed prenatally? MSAFP (maternal serum α-fetoprotein levels), fetal ultrasound, and amniocentesis (for α-fetoprotein and acetylcholinesterase)1 True or false: With proper and adequate treatment, most myelomeningocele patients survive at the outset. True1 What conditions are commonly associated with myelomeningocele? • Hydrocephalus (80–90%) • Chiari II malformation (approximately 100%) • Tethered cord syndrome (one third require de-tethering) • Hydrosyringomyelia = hydromyelia + syringomyelia (50–80%)1 What conditions most commonly contribute to mortality in myelomeningocele patients? Infant Chiari malformation and unresolved shunt obstruction later in life1 How is intelligence quotient affected in myelomeningocele patients? Intelligence is affected by the presence or absence of hydrocephalus and to the level where the myelomeningocele is occurring. Shunt infections further worsen the IQ.1 A lesion above which level is predictive of poor ambulation in patients with motor paralysis? L31 Urinary social continence is attained in what percentage of school-age children? 80–85%1 What procedures (besides correction of the actual lesion) will many myelomeningocele patients require? • Shunt for hydrocephalus (80–90%) • Tethered cord release • Chiari decompression • Orthopedic procedures (64% need at least one)1 In which cases can treatment be withheld? When there are coexisting potentially fatal congenital malformations or chromosomal anomalies1 What are the three phases of myelomeningocele management? 1. Stabilization and closure of the lesion 2. Correction of hydrocephalus, if needed 3. Lifelong management of shunt malfunctions, Chiari malformation, tethered cord, and syringomyelia1 How should the patient be assessed prior to surgical treatment? • Determine functional level of lesion via spontaneous motor movement and pinprick stimulation • Look for signs of hydrocephalus • Evaluate brainstem function • Genetics evaluation if there is evidence of chromosomal abnormalities, genetic syndromes, or other lethal malformations1 Within the first 48 to 72 hours of life1 What does surgical correction of myelomeningocele entail? • Dissection of the placode with sparing of underlying nerve roots and feeding and draining vessels • Section of filum terminale if present to reduce risk of tethering • Re-creation of the neural tube by bringing the edges of the placode together pia to pia • Elevation of the surrounding dura to form a spacious dural sac (avoid constriction of newly closed cord)1 What is the postoperative status? Stable neurological, orthopedic, and urological deficits, which is the best possible outcome1,2 What about radiographic findings? Radiographic abnormalities almost always present, so the findings must be compared with the patient’s baseline to catch and correct any deterioration (check for shunt malfunction first!).1 Can in-utero surgical correction be done? Yes, it is done in some tertiary centers and may reduce the severity of Chiari II and neurological problems.2 To what are 20 to 65% of myelomeningocele patients allergic? Latex or products containing latex1 What is the No. 1 lifelong neurosurgical problem for myelomeningocele patients? Maintenance of correct shunt functioning1 What is occult spinal dysraphism (OSD)? A set of disorders resulting from abnormalities in embryological development of midline dorsal neural, mesenchymal, and cutaneous ectodermal structures What other term is often used interchangeably with OSD? Spina bifida occulta What specific disorders does OSD include? • Diastematomyelia (split cord malformation type I) and diplomyelia (split cord malformation type II) • Meningocele manqué (dorsal bands) • Dermal tract/sinus ± intraspinal tumors • Lipoma of conus ± filum • Abnormal filum (e.g., tight filum terminale → tethered cord syndrome) What do all of the above disorders have in common? • Spinal cord tethering, neural compression, and myelodysplasia are the main perpetrators of neurological problems. • Surgical treatment can ameliorate progressive worsening of neurological problems, but most procedures likely will not reverse static deficits. • In cases of more complex malformations, postoperative re-tethering is likely, so it is imperative to monitor such patients for any deterioration (neurological, urological, orthopedic). Is the incidence of OSD known? No; it is not as obvious as open spinal dysraphism (such as anencephaly or myelomeningocele) so one must look for skin markings, neurological problems, and musculoskeletal deformities. What causes a tight filum terminale? Absence of retrogressive differentiation and tissue response to remaining pluripotent cell rests The appearance of what structures is characteristic of tight filum terminale? Fat, collagenous bundles, and vessels What are the two types of split cord malformations? • Type I (diastematomyelia) • Type II (diplomyelia) What causes split cord malformation (both types)? Persistent atypical neurenteric canal separating the yolk sac and the amnion What is meningocele manqué? Failed development of a meningocele due to spontaneous healing or scarring • Manqué means “missing” in French How is meningocele manqué usually described as? A dorsal band (fibrous tissue, can have meningeal components) ± accompanying nerve roots/dorsal root ganglia How is meningocele manqué usually detected? Incidentally during surgical operation for associated dysraphic lesions What is the most common type of OSD? Lipoma of caudal neural structures (filum and conus) How do all lipomas of the filum and conus appear histologically? Normal adipose tissue with interspersing collagenous bands ± muscle, nervous tissue, cutaneous end organs, tissues neuroectodermal in origin From which normal structure should these lipomas be distinguished? Sacral fat pad (limited to the sacrum) Lipomas of the conus usually run between which spinal levels? L5 → S3 How do lipomas of the conus form? Early (before closure of neural tube) dysjunction of cutaneous and neural ectodermal layers and differentiation of mesenchymal cells (that had migrated dorsally) into fatty, mesenchymal, and neuroectodermal tissues What are some of the more complex forms of lipomas of the conus? • Lipomyelomeningocele • Lipomyelocystocele • Lipomyelocele How do these complex forms differ from other types of OSDs? They inflict mass effect on and within the distal spinal cord. Where do dermal sinuses usually occur? Midline lumbosacral junction How do they (and their associated tumors) arise? Partial dysjunction of neural ectoderm and epithelial ectoderm True or false: Dermoid tumors are seen in 30% of all patients with dermal sinus–associated tumors. False; dermoid tumors are seen in approximately 80% of patients, whereas epidermoid tumors are seen in approximately 20% of patients. Are intraspinal teratomas seen in patients with dermal sinuses? Yes, but rarely Where do the majority of dermal sinuses terminate? At the filum terminale or conus medullaris What is a terminal syrinx? A cystic dilation usually placed above other OSD anomalies in the lower third of the spinal cord In how many cases of OSD is a terminal syrinx present? One third of cases Does the presence of a terminal syrinx indicate symptomatology? Usually. In two thirds of such cases there will likely be pain, motor weakness, and bladder and bowel problems. With what condition is terminal syrinx commonly associated? Tight filum terminale Which syndromes are associated with OSD? • Caudal agenesis/caudal regression syndrome • Vertebral anomalies such as sacral dysgenesis and abnormalities of laminae, vertebral bodies, pedicles, etc. What are the main factors contributing to neurological deficits in OSD? • Intrauterine developmental malformation of neural structures • Compression or expansion in the conus medullaris due to lesions (e.g., lipoma of the conus or tumors) • Traction on radicular and medullary vasculature, spinal cord, and nerve roots by such lesions resulting in tethering of these structures to the neighboring spinal canal, dura, or extradural tissues • Meningitis, abscess, hemorrhage What are the clinical signs and symptoms of OSD? Midline cutaneous stigmata (80%) • Hypertrichosis: “faun’s tail,” especially in split cord malformation • Atretic meningocele: remnant of fetal meningocele (can see bluish tint of CSF underneath thin skin) • Dermal sinus: patent skin-lined dimple leading to the dura or spinal cord (easy route for bacterial infection) • Subcutaneous lipoma (usually at lumbosacral junction) • Dermal appendages: ranges from simple (tail-like structures) to complex (accessory limbs) • Capillary hemangioma/nevus: indicative of underlying dysraphism when in combination with other cutaneous stigmata Orthopedic deformities • Foot and leg deformities: foot deformity is most common orthopedic sign in OSD • Scoliosis: in 90% of split cord malformation cases and 25% of tight filum terminale cases Urological deficits • Neurogenic bladder • Urinary tract infections • Incontinence Neurological signs and symptoms • Infants • Toddlers • Older children • Young adults • All ages What diagnostic studies are done for assessment of OSD patients? • MRI for diagnosing and monitoring • Mnemonic: “T2 shows H2O” • AP and lateral plain radiographs to assess for vertebral defects and for surgical planning • Ultrasound for screening of asymptomatic infants with sacral dimples and isolated strawberry hemangioma and for assessment of myelocystocele • Electrophysiological techniques for intraoperative monitoring of functioning neural tissue • Urodynamic recording for monitoring of bladder function What can surgical treatment not correct? Congenital myelodysplasia and already present/static clinical deficits Which OSD-associated conditions are easily and safely surgically correctable? Dermal sinuses, dermoid tumor, terminal and filar lipomas, split cord malformation, meningocele manqué, and neurenteric cysts Why are complex OSDs associated with difficult surgical decision making? Because their natural histories are lesser known and unpredictable What are the key surgical treatment steps for a tight filum terminale? • S1 → midsacrum laminectomy/laminoplasty • ID of filum through dural and arachnoidal opening • Separation of filum from surrounding nerve roots • Coagulation and division of filum What are the key surgical treatment steps for split cord malformation? Type I: • Dissection of median septum from dura in subperiosteal plane • Extradural resection of septum (beware of blood vessels) • Opening of dural sleeves → separate hemicorddural adhesions • Resection of medial dura Type II (less challenging): • Exploration along entire length of split • Disconnection of fibrous septum from ventral dura What is the key surgical treatment step for meningocele manqué? Cutting of the bands if they run dorsally (not ventrolaterally) to let the cord relax What are the key surgical treatment steps for lipomas of the conus? • Skin excision above and below the subcutaneous lesion • One-level laminectomy on caudal-most intact lamina • Exposure and opening of the dural-lipoma junction, taking care to avoid nerve roots • Separation of dural edge from the neck of lipoma • Debulking of the intramedullary area of the lipoma • Approximation of edges of spinal cord to minimize chances of re-tethering • Exploration of other cord abnormalities • Division of filum terminale • Dural watertight closure Fig. 73.1 Sagittal T2-weighted image of lipomyelomeningocele. Note how intensely hyperintense the fat appears on MRI (arrows).
Neural Tube Defects
73.1 Myelomeningocele
 Waardenburg’s syndrome in humans
Waardenburg’s syndrome in humans
 Derangement of DNA synthesis because folate is integral in metabolic pathways of purine and pyrimidine formation
Derangement of DNA synthesis because folate is integral in metabolic pathways of purine and pyrimidine formation
 Exposure of maternal or fetal methionine synthase or 5,10-methylenetetrahydrofolate reductase mutations?
Exposure of maternal or fetal methionine synthase or 5,10-methylenetetrahydrofolate reductase mutations?
 Valproic acid (antiepileptic)1
Valproic acid (antiepileptic)1
 Distal continuation of spinal cord
Distal continuation of spinal cord
 Termination of spinal cord at the placode2
Termination of spinal cord at the placode2
 Almost always need revision eventually
Almost always need revision eventually
 Full fontanelle, split cranial sutures, enlarged head circumference
Full fontanelle, split cranial sutures, enlarged head circumference
73.2 Occult Spinal Dysraphism1
 Two dural sheath-housed hemicords divided by an osteocartilaginous median septum
Two dural sheath-housed hemicords divided by an osteocartilaginous median septum
 One dural sheath enveloping two hemicords divided by a fibrous septum
One dural sheath enveloping two hemicords divided by a fibrous septum
 Lipoma + subcutaneous meningocele
Lipoma + subcutaneous meningocele
 Lipoma + terminal hydromyelia
Lipoma + terminal hydromyelia
 Extraspinal extension of cord without meningocele or terminal hydromyelia
Extraspinal extension of cord without meningocele or terminal hydromyelia
 Spectrum of sporadic congenital malformations caudally in the developing embryo
Spectrum of sporadic congenital malformations caudally in the developing embryo
 Composed of gastrointestinal, urological, orthopedic, and neurological anomalies
Composed of gastrointestinal, urological, orthopedic, and neurological anomalies
 → too much tension within cord, becomes ischemic and is exacerbated by movement
→ too much tension within cord, becomes ischemic and is exacerbated by movement
 Decreased spontaneous leg movement
Decreased spontaneous leg movement
 Absent reflexes
Absent reflexes
 Leg atrophy (hidden by baby fat)
Leg atrophy (hidden by baby fat)
 Foot asymmetry
Foot asymmetry
 Developmental delay (e.g., walking)
Developmental delay (e.g., walking)
 Abnormal gait
Abnormal gait
 Asymmetrical sensorimotor dysfunction
Asymmetrical sensorimotor dysfunction
 Painless foot burns
Painless foot burns
 Upper motor neuron signs (e.g., hyperreflexia)
Upper motor neuron signs (e.g., hyperreflexia)
 Back and leg pain
Back and leg pain
 Chronic or acute back and leg pain
Chronic or acute back and leg pain
 Spasticity and hyperreflexia
Spasticity and hyperreflexia
 Meningitis
Meningitis
 Paraplegia
Paraplegia
 T1-weighted imaging: anatomical detail of spinal cord and filum terminale, presence/absence of fat
T1-weighted imaging: anatomical detail of spinal cord and filum terminale, presence/absence of fat
 T2-weighted imaging: demonstrates tumors and water-containing structures (syrinxes, myeloceles)
T2-weighted imaging: demonstrates tumors and water-containing structures (syrinxes, myeloceles)
 Concurrent drainage of cystic cavity if lipomyelocystocele is present
Concurrent drainage of cystic cavity if lipomyelocystocele is present
Neural Tube Defects
Only gold members can continue reading. Log In or Register to continue





Full access? Get Clinical Tree


