Neurocutaneous Syndromes
Bernard L. Maria
John H. Menkes
The neurocutaneous syndromes are marked by the conjoined abnormalities of skin and nervous system. The term phakomatoses (phakomatosis from fakos, Greek for lentil) is reserved for a group of diseases in which the subject is predisposed to tumors of the skin, nervous system, and other organs. The major entities included among the phakomatoses are the neurofibromatoses, tuberous sclerosis (TS), Sturge-Weber syndrome (SWS), von Hippel–Lindau disease, ataxia-telangiectasia (AT), and hypomelanosis of Ito (HI).
Additionally, numerous other conditions exist, many of uncertain heredity and some extremely rare, in which abnormalities of skin are linked with those of the nervous system. These are detailed in a book edited by Gomez (1).
Though a common pathogenesis of all neurocutaneous syndromes has eluded investigators for a century because the various diseases in this category have very different clinical presentations, genetic transmissions, and pathological findings, a unifying etiology has recently been proposed. This suggests that all neurocutaneous syndromes are disorders of neural crest (i.e., neurocristopathies) that can affect all three primitive germ layers. The known genes that regulate neural crest formation, migration or terminal differentiation also serve as tumor-suppressor genes (e.g., NF1, TSC1,2), hence the high incidence of neoplasms, both benign and malignant, in these diseases (1a).
NEUROFIBROMATOSIS (VON RECKLINGHAUSEN DISEASE)
No longer considered to be a single disorder, neurofibromatosis has been divided into at least two genetically distinct forms. The common form, once known as peripheral neurofibromatosis, is called neurofibromatosis 1(NF1). The other, rarer form, once termed central neurofibromatosis, is now called neurofibromatosis 2(NF2). Additionally, several authorities distinguish segmental neurofibromatosis, in which the features of NF1 are confined to one part of the body; spinal neurofibromatosis, characterized by the late appearance of spinal cord tumors; and a condition marked by autosomal dominant café au lait spots. Oh and colleagues have recently reported that a subset of patients may meet diagnostic criteria for both NF1 and NF2 (2).
Neurofibromatosis 1
NF1 is characterized by multiple tumors within the central and peripheral nervous systems, cutaneous pigmentation, and lesions of the vascular system and viscera. Additionally, a tendency exists for a variety of tissues to undergo malignant transformation. Although it was described initially in the eighteenth century, and more succinctly in 1849 by Smith (3), von Recklinghausen in 1882 first combined the various features of the condition and termed it neurofibromatosis (4). The disease occurs in approximately 2 to 3 per 10,000 live births and is transmitted as a dominant trait with variable expression but virtually complete penetrance by the age of 5 years (5). It is the most common single-gene defect to affect the nervous system. Approximately one-half of the cases appear to be sporadic, and the mutation rate has been estimated at 1 in 10,000 gametes per generation, one of the highest mutation rates in humans (6). Stephens and coworkers found that 93% of new mutations were in the paternally derived chromosome (7). For as yet unknown reasons, no parental age effect occurs. As determined by linkage analysis and translocation breakpoints, the gene for NF1 is located on the long arm of chromosome 17, near the centromere (q11.2). It has been cloned and consists of 60 exons that are spread out over 350 kb of genomic DNA and gives rise to three alternative-spliced messenger RNA transcripts (8,9). NF1 is heterogeneous at the mutation level, with more than 300 independent mutations having been reported (10). The gene encodes a cytoplasmic protein, named neurofibromin, which contains a 2818 amino acid segment whose only demonstrated function is down-regulation of the Ras signal transduction. The peptide domain encoded by exons 21 to 27 activates the intrinsic guanosine triphosphatase of Ras proteins (N-, K-, and H-ras), which leads to hydrolysis of bound guanosine triphosphate and inactivation of downstream signaling. Neurofibromin inactivates the
tumor gene p21ras by stimulating its GTPase activity and converting the active form of p21ras into its inactive form. Inasmuch as the active form of p21ras is a specific growth regulator for astrocytes, the NF1 gene functions as a tumor-suppressor gene (11,12,13). This is confirmed by the observation that loss of NF1 gene expression occurs in at least some neurofibroma, in neurosarcoma, and in leukemic cells derived from NF1 subjects (11,14,15). Neurofibromin also has been shown to be associated with cytoplasmic microtubules in the brain and is believed to be involved in signaling within the central nervous system (CNS).
tumor gene p21ras by stimulating its GTPase activity and converting the active form of p21ras into its inactive form. Inasmuch as the active form of p21ras is a specific growth regulator for astrocytes, the NF1 gene functions as a tumor-suppressor gene (11,12,13). This is confirmed by the observation that loss of NF1 gene expression occurs in at least some neurofibroma, in neurosarcoma, and in leukemic cells derived from NF1 subjects (11,14,15). Neurofibromin also has been shown to be associated with cytoplasmic microtubules in the brain and is believed to be involved in signaling within the central nervous system (CNS).
The NF1 gene is large and is intrinsically hypermutable; more than 300 mutations have been described, and only rarely has the same mutation been identified in unrelated patients. Mutations include large deletions seen in 7.5% to 17% of patients (16,17), frame shifts, stop mutations, and point mutations. The majority (60% to 70%) of mutations result in the formation of truncated and nonfunctioning neurofibromin. Somatic mosaicism is fairly common; its exact frequency has not been ascertained (16). NF1 gene expression is complex and is modulated post-transcriptionally by numerous alternative splicings and RNA editing (18). Some of the alternative transcripts lack tumor suppressor activity and are developmentally regulated. Their role in producing the clinical phenotype of NF1 is not understood (9,18,19).
It, therefore, comes as no surprise that there is much variability in the expression of NF1, even within the same family. Since the NF1 gene was cloned in 1990, two major pathogenetic mechanisms have been considered: (a) loss of NF1 function as a tumor suppressor gene, and (b) heterozygous mutation of the gene leads to haploinsufficiency of the gene product (20). A detailed review of the pathogenesis is beyond the scope of this text. The interested reader is referred to a chapter by Huson and Korf (5).
Correlation between the genetic mutation and the clinical expression is still poor. However, a significant proportion of subjects with severe manifestations, including dysmorphic features, have large deletions in the NF1 gene (21).
Pathology
The most striking neuropathologic feature of neurofibromatosis 1 is the presence of tumors along the major peripheral nerves, with the ulnar and radial nerves being involved most frequently. Neurofibromas are the most common tumor type, but schwannomas also can be seen. Tumors that are prone to develop within the CNS include primarily optic gliomas; pilocytic astrocytomas of the third ventricle, cerebellum, and spinal cord; and high-grade astrocytomas (22,23). Additionally, neurofibromatosis has been associated with a number of other neoplastic processes with a greater than random frequency (24). These include leukemia, Wilms tumor, neuroblastoma, and pheochromocytoma (25). A syndrome of multiple endocrine neoplasia characterized by bilateral pheochromocytomas, medullary thyroid carcinoma, and multiple neuromas and café au lait lesions has been delineated (26). Although generally benign, both central and peripheral neurofibromas can undergo malignant degeneration. This is particularly likely to occur with the plexiform neurofibroma, for which the risk for malignant transformation to neurofibrosarcoma has been estimated at 5% (27).
Clinical Manifestations
NF1 is a progressive disease process that can affect almost every organ. When many peripheral lesions are present, few lesions tend to be within the CNS. The reverse also is true (28).
The most common skin lesions are the café au lait spots. These are numerous light brown areas, usually located over the trunk, with smooth, well-defined borders and uniform pigmentation. They are seen in virtually every patient with NF1, and they result from an aggregation of neural crest-derived pigmented melanoblasts in the basal layer of the epidermis (5). They are present at birth, and their number and size increase until puberty. According to Crowe and associates, at least six such lesions are necessary for a diagnosis of NF1 (29). There is no correlation between the number of spots and the severity of the NF1. Less frequent are diffuse freckling, freckling in the armpits and groin that tends to begin between 3 and 5 years of age, and large areas of faintly increased pigmentation (melanoderma). Although usually present before the onset of neurologic symptoms, these pigmentary abnormalities are not striking during infancy but intensify with age, particularly after puberty.
Various types of cutaneous tumors can be found (Fig. 12.1). The most characteristic for NF1 is the pedunculated molluscum fibrosum and the subcutaneous neurofibromas. The latter consist of an overgrowth of Schwann cells admixed with tortuous nerve fibers and perineural fibroblasts. The number of neurofibromas is highly variable, and they are located singly or in groups along nerve trunks. Generally, cutaneous tumors tend to enlarge slowly throughout life, and they occur at an unpredictable rate (20). Plexiform neurofibromas can occur in all affected tissues and lead to exophthalmos, or defects of the skull and orbit, or hypertrophy of one or more extremity, sometimes with overlying hyperpigmentation. Plexiform neurofibromas can be quite extensive and infiltrative and can be associated with soft tissue overgrowth. There is a risk that a neurofibroma will transform into a malignant peripheral nerve sheath tumor, which is associated with anaplastic changes and multiple mitoses. The lifetime risk for malignant peripheral nerve sheath tumor is estimated to be 5 to 10% (30).
Multiple nodules within the iris (iris hamartomas) (Fig. 12.2) were first described by Lisch (31). Lisch nodules are seen in almost all affected individuals aged 21 or more
years, but only in one-half of children aged 5 to 6 years (32). Initially light colored, these melanocytic hamartomas become darker with time. They do not affect vision, but they are helpful diagnostic markers (32).
years, but only in one-half of children aged 5 to 6 years (32). Initially light colored, these melanocytic hamartomas become darker with time. They do not affect vision, but they are helpful diagnostic markers (32).
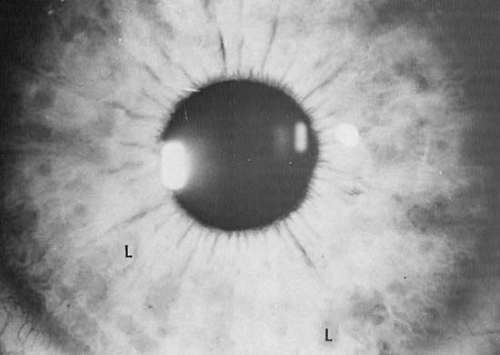 FIGURE 12.1. Lisch nodules of iris (L). (Courtesy of Dr. Bronwyn Bateman, Department of Ophthalmology, University of Colorado School of Medicine, Denver.) |
Short stature is common. It was seen in 31.5% of patients in the series of Huson and Korf (5). In some children, a number of risk factors, including suprasellar lesions and skeletal deformities are responsible. In addition, growth hormone deficiency can be common; Vassilopoulou-Sellin and coworkers found defective growth hormone levels in 79% of children who had no obvious medical or radiologic lesions to account for their short stature (33).
Macrocephaly is common, and 16% of children in Riccardi’s series (27), 45% of children in the series of Huson and Korf (5), and 38% of children in Young and coworkers (35) series had a head circumference at or above the 98th percentile (34). Only rarely is there associated hydrocephalus, typically owing to aqueductal stenosis. Various skeletal abnormalities not associated with tumors have been described. The most common location for dysplasia of a long bone is the tibia, although other long bones can be affected (36). Low cervical or thoracic kyphoscoliosis can result in a narrow angulation of the spine that may require surgery in 5% of patients (35,37). Scoliosis was noted in 32% of children in the series of Holt, and its incidence increases with age (38). Less commonly, one observes scalloping of the posterior portion of the vertebral bodies. This scalloping is caused by a dural ectasia, the
consequence of congenital weakness of the dura and the resulting pressure on the vertebral bodies. Anterior and lateral meningoceles, which are more common in adults, also result from the dural weakness. Bony rarefactions, the consequence of subperiosteal neurofibromas, can arise within the spine, the pelvis, particularly the iliac wings, or the skull. These rarefactions can induce pathologic fractures. Bony overgrowth, often with contiguous elephantiasis, is seen in approximately 10% of patients. Radiographic findings are reviewed by Holt (38), Klatte and coworkers (39) and Ippolito and coworkers (40).
consequence of congenital weakness of the dura and the resulting pressure on the vertebral bodies. Anterior and lateral meningoceles, which are more common in adults, also result from the dural weakness. Bony rarefactions, the consequence of subperiosteal neurofibromas, can arise within the spine, the pelvis, particularly the iliac wings, or the skull. These rarefactions can induce pathologic fractures. Bony overgrowth, often with contiguous elephantiasis, is seen in approximately 10% of patients. Radiographic findings are reviewed by Holt (38), Klatte and coworkers (39) and Ippolito and coworkers (40).
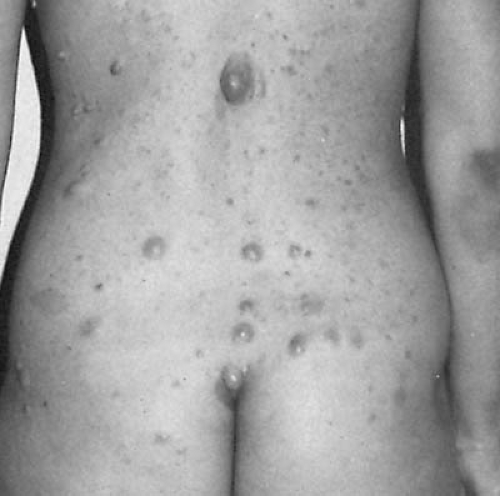 FIGURE 12.2. Neurofibromatosis. Posterior view demonstrating various types of cutaneous tumors. These include the pedunculated molluscum fibrosum and subcutaneous neurofibromas. Note the area of hyperpigmentation of the right elbow and a typical café au lait lesion. (Courtesy of Dr. V. M. Riccardi, Neurofibromatosis Institute, La Crescenta, CA.) |
Headache is common in children with NF1 and usually occurs in the absence of structural lesions (41). Severe migrainelike headache occurs in 20% to 25% of children with NF1, but because of the risk of CNS tumors, prompt neuroimaging with MRI is warranted. Hypertension can develop owing to the presence of a pheochromocytoma, which is seen in 1% to 4% of subjects. It also can be the result of renal artery stenosis, the most common of a variety of arterial abnormalities seen in neurofibromatosis (42,43). The microscopic picture of these arterial abnormalities is one of an intense subintimal proliferation of the spindle cells, which are believed to be of Schwann cell origin. Congenital heart disease has been reported in NF1 as well.
Cognitive Disabilities
Although as many as 30% to 60% of children with neurofibromatosis have learning disabilities, only a small proportion are severely retarded (27,34,46). Thus, in the series of Ferner and colleagues, only 8% of patients with NF1 had an IQ below 70 (47). All studies designed to investigate the cognitive deficits of NF1 subjects have shown a significant lowering in full-scale IQ when compared with unaffected siblings. As a rule, children tend to do better on verbal tasks rather than performance tasks and show deficits in visuospatial areas, attention, short-term memory, and reading (47,48). Both nonverbal and verbal learning problems occur, and the children may be easily distractible and poorly organized (49). Mental retardation is only slightly more common in patients with NF1 than in the general population (4% to 8%) (46). These deficits are believed to result from cortical heterotopias and other malformations of cerebral architecture such as glial nodules and other hamartomatous lesions (50) as well as from the presence of abnormal myelin. Studies conducted by Costa and Silva suggest that learning disabilities associated with NF1 are associated with excessive Ras activity that leads to increased γ-aminobutyric acid (GABA) inhibition and to decreased long-term potentiation (51).
Malformations have been demonstrated by magnetic resonance imaging (MRI) as small focal areas of increased signal (unidentified bright objects—UBOs) on T2-weighted scans in 60% to 70% of patients with neurofibromatosis (35,52). Areas of increased signal (isointense on T1-weighted images) are located with particular frequency in the globus pallidus, brainstem, optic tracts, thalamus, and cerebellum (53,53a). They exert no mass effect, do not enhance with contrast, and are not visible on CT scan; they are asymptomatic and unrelated to the presence of macrocephaly. UBOs tend to diminish or disappear over the years and are rare in subjects older than 30 years of age. In the experience of DiMario and Ramsby, lesions in the basal ganglia and cerebellum decrease in size and number over time, whereas lesions in the brainstem tended to increase in both number and size (54). Pathologic studies suggest that these hyper-intense areas on MRI may represent dysmyelination or increased water content in the brain. Jurkiewicz and colleagues have shown that proton magnetic resonance spectroscopy may distinguish UBOs from astrocytomas in NF1 (55). Studies present conflicting data as to whether the number of abnormalities seen on MRI correlate with the severity of cognitive deficits (49,56). The most recent series, published in 2003 by Feldmann and colleagues, suggest that as a rule, patients with focal areas of increased signal on T2-weighted MRI studies do worse on cognitive and fine motor performance than NF-1 patients who do not show these lesions (56a). When lesions are seen in the brainstem, they should not be confused with a neoplasm (50).
Intracranial Tumors
Children with NF1 are at risk for optic pathway gliomas and brainstem gliomas. In addition, there appears to be an increased risk for the occurrence of benign and malignant neoplasms in other locations (the cerebrum or cerebellum), ependymomas, meningiomas, PNET/MBs, and malignant schwannomas arising from the cranial nerves (57).
Optic Pathway Gliomas.
Intracranial tumors can arise at any time of life, the optic pathway being the most common and the earliest site of involvement (58). In the series of Holt, optic pathway gliomas were found in 23% of children with neurofibromatosis (38). This compares with an incidence of 15% in the series of Huson and Korf (5) 20% in the series of Poyhonen (58a), and 19% in the series of Listernick and colleagues (59). The tumor is benign and histologically corresponds to a pilocytic astrocytoma. It is more common in girls, with a female to male ratio of 2:1 (59). Approximately one-half of patients who harbor optic pathway tumors develop signs or symptoms, and the tumor can involve any portion of the optic pathway (60). Bilateral optic nerve gliomas are seen almost exclusively in NF1 (61). Although optic pathway gliomas are an incidental finding in the majority of children with NF1, these neoplasms occasionally enlarge to distort and compress local structures, causing decreased visual acuity, visual field cuts, afferent papillary defect, decreased color vision, proptosis, strabismus, papilledema, optic nerve atrophy, and optic disk pallor. Precocious puberty is seen in
approximately 40% of subjects and results from compression of the hypothalamus and interference with the tonic central nervous system inhibition of the hypothalamic-pituitary-gonadal axis. The presence of precocious puberty in a child with NF1, therefore, should always arouse the suspicion of an enlarging chiasmatic glioma. The diencephalic syndrome also may be seen but is much more common in hypothalamic tumors in patients without NF1 (62).
approximately 40% of subjects and results from compression of the hypothalamus and interference with the tonic central nervous system inhibition of the hypothalamic-pituitary-gonadal axis. The presence of precocious puberty in a child with NF1, therefore, should always arouse the suspicion of an enlarging chiasmatic glioma. The diencephalic syndrome also may be seen but is much more common in hypothalamic tumors in patients without NF1 (62).
Decreased visual acuity is rarely a presenting complaint in children, even though it can be demonstrated by examination. The natural history of these optic pathway tumors in subjects with NF1 is not known, and their growth rate differs considerably from one patient to the next. In the series of Listernick and colleagues (59), no tumor growth was seen as determined by MRI over a mean interval of 2.4 years. Only a small proportion of intraorbital tumors progress, whereas tumors that involve the optic chiasm are more likely to progress. The consensus statement from the NF1 Optic Pathway Glioma Task Force has concluded that MRI screening of children with NF1 for optic pathway gliomas has only limited value, and even though asymptomatic tumors are often found, these only rarely progress. Riccardi believes that if an MRI study does not demonstrate an optic glioma by one year of age, these studies will remain negative (62a). Optic pathway gliomas also have been noted to spontaneously regress in patients with and without NF1, providing further evidence of their benign nature. Serial visual acuity examinations in asymptomatic children with NF1 under age 6 years and in symptomatic children are preferable and less costly than repeated MRIs. Accelerated linear growth or the appearance of premature secondary sexual characteristics should prompt an immediate work-up. Listernick and colleagues recommend that even for stable lesions, an MRI should be performed at 3, 9, 15, 24, and 36 months after diagnosis. Ophthalmologic exams should be performed at 3, 6, 12, 18, 24, and 36 months (63).
It is unusual for optic pathway gliomas to become aggressive, and tumors may even regress though they are symptomatic. One must carefully weigh the risks and benefits of any intervention in these children, attempting to preserve visual function while causing the least amount of harm. Treatment options include surgery, radiation, and chemotherapy, alone or in combination (23). The benefits of radical versus conservative surgery in these tumors have been debated in the literature (64). Radiotherapy has been shown to halt tumor progression, but there has been concern that it could transform the tumor to a higher-grade glioma and cause a vasculopathy (63). Reservations regarding the dangers of surgical resection and the potential toxicities of radiation therapy have led to the use of chemotherapy in progressive chiasmatic/hypothalamic gliomas, as reviewed by Rosser and Packer (23).
Focal or generalized seizures can appear early in childhood and were seen in 7% of patients in the series of Huson and Korf (5). Some of these patients had an electroencephalographic picture consistent with hypsarrhythmia. Because a significant proportion of patients with seizures and neurofibromatosis are ultimately found to have intracranial tumors, a child with cutaneous neurofibromatosis and seizures should be suspected of harboring a tumor and should receive imaging studies.
Brainstem Gliomas.
The true incidence of brainstem gliomas in NF1 is difficult to estimate because they are often mistaken for brainstem and cerebellar UBOs (65). Brainstem gliomas in NF1 cause neurologic symptoms such as headaches, hydrocephalus, and cranial neuropathies in 88% of patients. The medulla represents the primary tumor site in 80% of patients. Patients may require cerebrospinal fluid diversion, but unlike brainstem glioma in non-NF1 patients, radiotherapy or chemotherapy is rarely required. In fact, such tumors behave much like optic pathway gliomas in NF1 in that they have a relatively benign course and can regress spontaneously after becoming symptomatic. Pollack and colleagues reviewed the course of 21 children (mean age 9.5 years) with NF1 and brainstem gliomas (66). Twelve patients (57%) had clinically symptomatic lesions, with cranial neuropathies and hydrocephalus as most common symptoms. Only 4 children required specific intervention such as biopsy, resection, or adjuvant radiation. All children were alive at the time of the report, and radiographic progression was seen in only 9 children (3 clinically deteriorated).
Tumors of the Peripheral Nerves
Tumors of the peripheral nerves can arise at any age and can involve any of the major nerves. Even though these tumors are occasionally painful, surgical removal must be weighed carefully against the possibility of the procedure producing considerable neurologic deficit. Malignant degeneration of neurofibromas occurs in less than 3% of children, but appears more frequently in adults (27). Tumors also can arise within the autonomic nerve supply of various viscera. According to Kissel and Schmitt, the stomach, tongue, mediastinum, large intestines, and adrenal medulla are the most common sites (67). Treatment options for patients with progressive plexiform neurofibromas have been limited, with surgery as the only proven modality. Trials that have evaluated antihistamines, maturation agents, and antiangiogenic agents have had mixed results that are difficult to interpret, and therapy is moving toward a more biologically based approach (68).
Intraspinal Tumors
Intraspinal tumors are generally slower to develop than intracranial tumors, and asymptomatic spinal cord tumors are commonly detected on routine neuroimaging studies. The youngest patient with a symptomatic intraspinal tumor in Canale’s series was 20 years of age (44). Approximately one-half of intraspinal tumors are multiple, and occasionally, they are accompanied by malformations
such as syringomyelia. Familial spinal neurofibromatosis is a variant of NF1. The condition is marked by the development of multiple spinal cord tumors during adult life (67,69,70).
such as syringomyelia. Familial spinal neurofibromatosis is a variant of NF1. The condition is marked by the development of multiple spinal cord tumors during adult life (67,69,70).
TABLE 12.1 Neurological Complications in Patients with NF-1 | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
Cerebral Infarction
Cerebral infarctions are common and can be responsible for the abrupt evolution of neurologic signs. They result from cerebrovascular occlusive disease and most commonly affect the supraclinoid portion of the internal carotid artery or one of its major branches (71,72). More than one-half of the patients with occlusive disease of the internal carotid have the arteriographic picture of moyamoya disease (71,72).
The incidence of some of the neurologic complications encountered in NF1 and NF2 is presented in Table 12.1 (58a,72a).
Several conditions are related to NF1. Watson syndrome—characterized by dominantly transmitted pulmonary valve stenosis, café au lait spots, and low to normal intelligence—is believed to be allelic with NF1 (73). The concurrence of Noonan syndrome with NF1 may represent either a contiguous gene syndrome or the coincidental segregation of two autosomal dominant conditions (see Chapter 4 (73a)).
Diagnosis
Despite the advances in understanding the molecular biology for NF1 and NF2, the diagnosis for both conditions is still largely based on clinical criteria. The diagnosis of NF1 cannot be made with certainty before 1 year of age in almost half of affected children with a negative family history (74). The appearance of most signs of NF1 is age dependent, and reliability of diagnostic criteria improves as the child grows older (75). At this time, expert consensus does not support the use of UBOs as a diagnostic criterion because adding it the the National Institutes of Health (NIH) Diagnostic Criteria does not improve their sensitivity significantly (74a). The NIH diagnostic criteria for NF1 are two or more of the following:
Six or more café au lait macules whose greatest diameter is more than 5 mm in prepubertal patients and more than 15 mm in postpubertal patients
Two or more neurofibromas of any type, or one or more plexiform neurofibroma
Freckling in the axillary or inguinal region (Crowe’s sign)
An optic pathway tumor
Two or more Lisch nodules (iris hamartomas)
A distinctive osseous lesion such as sphenoid wing dysplasia or thinning of the cortex of the long bones (with or without pseudoarthrosis)
At the moment, DNA-based testing is unnecessary to make a diagnosis if the NIH criteria are met. DNA testing for the diagnosis of NF1 is limited because present techniques detect only approximately 70% of mutations (5), and detection of a specific mutation does not predict the severity of the disease. Although a solitary café au lait spot can occur in the normal population, the incidence of more than four such lesions in nonaffected persons is low, and in the absence of other symptoms of neurofibromatosis, the lesions can indicate a partial penetrance of the disease (80). Conversely, some 75% of individuals with proven NF1 have six or more café au lait spots 1 cm or more at the largest diameter (29).
Both parents should be examined with particular attention to the presence of café au lait spots, subcutaneous neurofibromas, and Lisch nodules. Detection of Lisch nodules often requires slit-lamp examination by an ophthalmologist. If one parent has the stigmata of NF1, the condition in the offspring is not a new mutation, and a 50% chance exists for it to occur in each subsequent sibling. The risk for the patient’s own potential offspring is the same. If neither parent has any abnormalities, a new mutation is presumed, and the recurrence risk for NF1 is no greater than in the general population. Prenatal diagnosis of the condition can be made by linkage analysis, if two or more family members are affected (79).
Treatment and Prognosis
Therapy is symptomatic. Most pediatric patients with NF1 should be seen in a multispecialist clinic at intervals of at least 6 months to 1 year to detect and manage the various potential complications. The necessity or value of routine cranial MRI scans is a matter of debate because it is becoming apparent that the detection of asymptomatic lesions may not alter clinical management (79). However,
the detection of asymptomatic optic pathway gliomas may alter the intensity of ophthalmologic monitoring. When tumors are confined to peripheral nerves, the long-term prognosis is generally good. The prognosis for intracranial tumors depends on their location and whether they are single or multiple. In a follow-up study of patients with NF1 first reported in 1951, Sorensen and coworkers found that survival was limited by an incidence of neoplasms that was four times greater than seen in the normal population. Thus, 84% of patients developed a glioma, and second tumors were seen five to eight times more frequently than expected. Malignancies were encountered in one-third of the cohort, with female subjects having a higher incidence than male subjects. Second neoplasms were seen in 83% of patients with optic gliomas and in 43% of patients with other types of gliomas (81). NF1 is a progressive disease, and more manifestations are usually present in older patients. The clinical variability and natural history of the burden of disease in NF1 has been reviewed by Friedman (75) and Young, Hyman, and North (35).
the detection of asymptomatic optic pathway gliomas may alter the intensity of ophthalmologic monitoring. When tumors are confined to peripheral nerves, the long-term prognosis is generally good. The prognosis for intracranial tumors depends on their location and whether they are single or multiple. In a follow-up study of patients with NF1 first reported in 1951, Sorensen and coworkers found that survival was limited by an incidence of neoplasms that was four times greater than seen in the normal population. Thus, 84% of patients developed a glioma, and second tumors were seen five to eight times more frequently than expected. Malignancies were encountered in one-third of the cohort, with female subjects having a higher incidence than male subjects. Second neoplasms were seen in 83% of patients with optic gliomas and in 43% of patients with other types of gliomas (81). NF1 is a progressive disease, and more manifestations are usually present in older patients. The clinical variability and natural history of the burden of disease in NF1 has been reviewed by Friedman (75) and Young, Hyman, and North (35).
Neurofibromatosis 2
NF2 is genetically and clinically distinct from NF1. It is far less common, with an estimated incidence of 1 in 33,000 to 40,000, and it is characterized by the development of CNS tumors, notably bilateral vestibular schwannomas (77). The gene for NF2 has been mapped to the long arm of chromosome 22 (22q11) and has been cloned. Its gene product, merlin (schwannomin), shares significant homology with several actin-associated proteins (78). Merlin is localized to the cell membrane and is believed to act as a membrane-cytoskeletal linker. It serves as a tumor suppressor by playing a role in the regulation of cell-cell adhesion and in the reorganization of the actin cytoskeleton in response to growth factors, confluency, and changes in the shape of the cell (82,83). Merlin is widely expressed in human brain; it is absent from almost all schwannomas and from many meningiomas and ependymomas isolated from subjects with NF2 (83a). A large number of gene mutations have been documented. Some 90% of patients have gross truncations of merlin as a result of nonsense or frame-shift mutations (84). These patients tend to be younger at onset of symptoms and at diagnosis and tend to harbor a large number of tumors (85).
Clinical Manifestations
In contrast to NF1, clinical manifestations and age of onset are similar within a given family, but differ considerably between families (86). The clinical manifestations of NF2 are highlighted by the presence of bilateral vestibular schwannomas (acoustic neuromas), which become manifest in more than 95% of genetically affected subjects (87). Generally, these tumors become symptomatic at puberty or thereafter. In addition, schwannomas occur in the other cranial nerves and the spinal and cutaneous nerves. Other tumors of the CNS seen in this condition include cranial and spinal meningiomas and multiple tumors of glial and meningeal origin. These tumors are readily detectable by imaging studies, with the acoustic neuromas appearing as a mass in the cerebellopontine angles or enlargement of the gasserian ganglia (52,88). As a rule, the mean age of onset of symptoms is in the second decade of life. In the series of Mautner and colleagues it was 17 years, with the age ranging from 2 to 36 years (88). In the same series, 44% of patients presented with deafness. Café au lait lesions were present in only 43% and in this series as in others they rarely number more than six (89,90). Cataracts (posterior subcapsular or cortical) were seen in 81%, and seizures were presenting complaints in 8%. Peripheral nerve tumors were seen in 68%. These are predominantly schwannomas, but also can be neurofibromas (89). These appear as discrete, well-circumscribed slightly raised lesions with a roughened, slightly pigmented surface. Other skin lesions such as nodular tumors or neurofibromas also are less common than in NF1. According to Riccardi, acoustic neuromas and optic glioma never coexist in a patient (27). The various neurological complications are summarized in Table 12.2 (90a).
TABLE 12.2 Neurological Complications in Patients with NF-2 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
Diagnosis
As is the case for NF1, the diagnosis of NF2 rests on clinical grounds. The criteria for NF2 are one or more of the following conditions:
Bilateral eighth nerve masses (vestibular schwannomas) seen with imaging techniques
A parent, sibling, or child with NF2 and either unilateral eighth nerve mass or any two of the following conditions: neurofibroma, meningioma, glioma, schwannoma, or juvenile posterior subcapsular lenticular opacity (76,89,91)
Patients with unilateral vestibular schwannomas and cataracts, or meningioma, glioma, or schwannoma are suspect for NF2, as are patients with multiple meningiomas plus unilateral vestibular schwannoma, cataracts, or glioma (89).
In 10% of cases of NF2, there is an identifiable mutation in merlin; for the remainder of patients, prenatal diagnosis requires a linkage study using DNA derived from at least two affected family members, if these are available.
Tuberous Sclerosis
Although the earliest report of a patient with TS is said to have been made by von Recklinghausen in 1863 (92), its first complete, albeit mainly pathologic, description is attributed to Bourneville, who, in 1880, was the first to call it TS (93). This is a protean disorder, chiefly manifested by mental deficiency, epilepsy, and skin lesions. It occurs with a frequency of 1 in 6,000 to 9000 and is transmitted as an autosomal dominant gene (94,95,96,97). Approximately one-third of cases have inherited a mutated TS gene from one of the parents; the rest are new mutations.
TS is genetically heterogeneous, with loci on chromosome 9q34.3 (TSC1), and 16p13.3 (TSC2) near the mapped location of the adult polycystic kidney disease gene (APKD1). Each locus accounts for approximately 50% of familial cases (96). The phenotypic expression of the two genetic defects appears to be similar, aside from the observation that TSC2 patients appear more prone to neurologic problems. TSC1 codes for hamartin, a 130-kd protein with no significant homology to any other known vertebrate protein (98). TSC2 codes for tuberin, a 200-kd protein, which functions as a tumor-suppressor gene (99). It acts as a GTPase activator for rap1, which is an effective proliferation signal, expressed in several tissues, notably astrocytes. Rap1 also is involved in morphogenesis and cell migration (100). Tuberin is most abundant in cerebral gray matter and increases during prenatal and postnatal development (101). The protein also may be involved in neuronal differentiation (102). The similarity in phenotypes produced by mutations in the TSC1 and TSC2 genes suggests that the genes somehow function together, and direct interaction between the two proteins has been shown (103). Hamartin and tuberin associate physically in vivo, and inactivation of either is believed to prevent the formation of a functional protein complex that regulates cell growth and proliferation (104,105). Nearly 1,000 mutations have been discovered to date, and genotype/phenotype correlation studies could provide guidance for optimal medical care in affected individuals. Relevant animal models, including conventional and conditional knockout mice, are valuable tools for studying the normal functions of tuberin and hamartin and how disruption of their expression gives rise to the variety of clinical features that characterize TS.
Mutations in the TSC2 genes are more readily detected in sporadic than in familial cases (106). Penetrance is variable. No family with two or more affected offspring has been encountered in which one parent did not have adenoma sebaceum or some other skin lesion characteristic for TS (107). Conversely, the risk of having more than one affected child is low when both parents are clinically unaffected because under such circumstances, the condition is probably a new mutation.
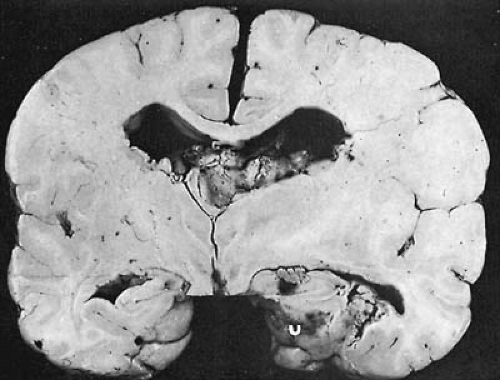 FIGURE 12.3. Tuberous sclerosis. A large intraventricular tuber produces increased intracranial pressure, flattening of the gyri, and herniation of the right temporal uncus (U). (Courtesy of Dr. P. Cancilla, Department of Pathology, University of California, Los Angeles, UCLA School of Medicine.) |
Pathology
Abnormalities can be found in the brain, eyes, skin, kidneys, bones, heart, and lungs. In the brain, three types of abnormalities occur: cortical tubers, subependymal nodules, and disorders of myelination. The most characteristic gross abnormality is the presence of tubers. These are numerous hard areas of gliotic tissue of varying size, after which this condition is named. Tubers can be located in the convolutions of any part of the cerebral hemispheres (Fig. 12.3). Less commonly, they are in the cerebellum, brainstem, or spinal cord. The highest frequency of tubers is in the frontal lobes, but the highest density is in the parietal regions (108). On histologic examination, tubers are sclerotic areas that consist of an overgrowth of atypical giant cells that exhibit cytomegaly (109) and express both gial and neuronal markers. Adjacent to these giant cells are dysplastic neurons that are characterized by aberrant dendritic arborizations and a dysmorphic cell body. Tubers may be dynamic lesions characterized by populations of cells undergoing proliferation, migration, and death. Crino recently showed that there is cell-specific activation of the mTOR/p70-S6 kinase/ribosomal S6 cascade in tubers and that giant cells express activated (phosphorylated) p70-S6-kinase and ribosomal S6 protein (110). The tuberin/hamartin complex regulates mTOR, and Rheb (Ras homologue enriched in brain), a Raslike GTPase, has been identified as a target of tuberin GAP activity (111,112,113,114,115,116). These findings support impaired hamartin/tuberin–mediated mTOR pathway and Rheb regulation.
Tubers likely form a constitutive activation of mTOR cascade during brain development as a consequence of impaired hamartin (TSC1) or tuberin (TSC2) function.
Tubers likely form a constitutive activation of mTOR cascade during brain development as a consequence of impaired hamartin (TSC1) or tuberin (TSC2) function.
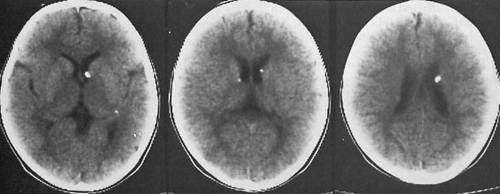 FIGURE 12.4. Noncontrast computed tomographic scan of tuberous sclerosis, taken at several levels, shows typical calcified subependymal tubers at the margins of the lateral ventricles and projecting slightly into the ventricles. (Courtesy of Dr. Hervey D. Segall, Children’s Hospital of Los Angeles.) |
Rapamycin, a specific inhibitor of mTOR, is currently in clinical trials and may prove useful in some TS-related tumors, including those that affect the brain. Similarly, farnesyltransferase inhibitors may prove useful in disrupting Rheb activation (117). Blood vessels in sclerotic regions show hyaline degeneration of their walls. In approximately one-half of subjects, calcium is deposited within the gliotic areas to an extent as to be visible on plain radiography of the skull or on computed tomographic (CT) scanning (Fig. 12.4). Subependymal nodules are found in the caudate nucleus and the ventricular walls, particularly in the region of the foramen of Monro. Tuber and nodule volumes are significantly positively correlated. Subependymal nodules are multiple small, tumorlike nodules that project into the ventricles and that because of their appearance on pneumoencephalography were described as “candle drippings.” Calcification of these nodules is common and increases with age. Subependymal giant cell astrocytomas arise from subependymal nodules, particularly in the area surrounding the foramen of Monro, and transitions between gliosis and astrocytomas are common. Their incidence in TS is approximately 10% to 15% (118). Although only rarely malignant, they often obstruct the foramen of Monro. It is of note that approximately one-half of high-grade and low-grade sporadic adult astrocytomas show reduced or absent expression of tuberin (119). In the remainder, the majority have an increased expression of rap1.
Myelination usually is diminished in the gliotic areas within and surrounding the cortical tubers. In addition, islets occur consisting of heterotopic cells within white matter. These are distributed in a linear pattern that follows the normal migratory path of primitive neurons between the germinal layer of the ventricles and the cortical surface.
Tumors also can arise from various viscera. In the heart, the characteristic lesion is the rhabdomyoma. The incidence of these tumors in children with TS can be as high as 50%. Characteristically, they are multiple and well circumscribed. Rhabdomyomas cause as many as one-fourth of infants to die from circulatory failure during the first few days of life, well before developing other stigmata of TS. Between 50% and 80% of patients develop multiple renal tumors, which are usually benign and of mixed embryonal type. Lungs are rarely involved, but when lesions are present in the lungs, they are usually cystic or fibrous. Other organs can be the seat of fibrocellular hamartomas (1). The pathologic features of the disease are extensively reviewed by Bender and Yunis (120).
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


