SUMMARY
Syndromes with neurodegeneration with brain iron accumulation (NBIA) are a group of neurodegenerative disorders characterized by abnormalities in brain iron metabolism with excess iron accumulation in the globus pallidus (GP) and to a lesser degree in the substantia nigra and sometimes adjacent areas. NBIA syndromes clinically present as neurodegenerative diseases with progressive hypo- and/or hyperkinetic movement disorders and a variable degree of pyramidal, cerebellar, peripheral nerve, autonomic, cognitive and psychiatric involvement, and visual dysfunction. Several causative genes underlying NBIA have been identified, which implicate abnormalities in mitochondrial function, lipid metabolism, and autophagy. Rapid developments prompted the review of this interesting field.
INTRODUCTION
Iron plays a key role in the metabolism of organisms, and its precise regulation is pivotal as both iron deficiency and iron overload may cause diseases of various kinds, including hematologic and neurologic disorders. In the brain, deposition of iron may lead to disturbance of cell function and development of neurologic symptoms. The basal ganglia appear particularly vulnerable to increased iron accumulation. In recent years, a growing number of conditions with excess iron accumulation in the GP and to a lesser degree in the substantia nigra and sometimes adjacent areas have been grouped together as syndromes with NBIA. NBIA syndromes share the clinical characteristics in that they present with a progressive hypo- and/or hyperkinetic movement disorder (with additional other features) and excessive iron deposition in the brain detectable by MR neuroimaging. Yet, several distinct forms of NBIA syndromes have now been molecularly defined and continue to be unraveled. Iron accumulation may also be demonstrated postmortem by brain histology, in association with degeneration of both neurons and astrocytes. Furthermore, spheroid bodies are common to many forms of NBIA, and there is overlap with the classification of neuroaxonal dystrophies. The pathophysiologic mechanisms are not fully understood but may differ between the various subtypes, but mitochondrial dysfunction, abnormal lipid metabolism, and altered autophagy have been implicated. The following chapter shall introduce and provide an overview of the different NBIA syndromes in order to facilitate the clinical diagnosis and therapeutic decisions (Fig. 23.1).
HISTORICAL MILESTONES: DISEASE DESCRIPTION AND GENE IDENTIFICATION
The first explicit description of an iron accumulation disorder dates back to the 1920s; brain histology revealed high levels of brain iron and spheroid bodies in a patient affected by a progressive extrapyramidal disorder (onset around 7–9 years, death in the early 20s) (1). Authors were Julius Hallervorden and Hugo Spatz, and hence, for many decades the term “Hallervorden–Spatz disease” was used but has now largely been replaced by disease names referring to the molecular (see below) underpinnings. In fact, in the old literature which dates to prior the gene identification, patients with different subforms of NBIA were probably published under the umbrella term of “Hallervorden–Spatz disease,” and its interpretation with regard to the different subtypes is thus limited. The new terminology tries to follow a pattern in that the first word(s) (or letters in the abbreviated name) reflect the molecular mechanisms and the last two words stand for “associated neurodegeneration,” that is pantothenate kinase–associated neurodegeneration (PKAN), PLA2G6-associated neurodegeneration (PLAN), etc.
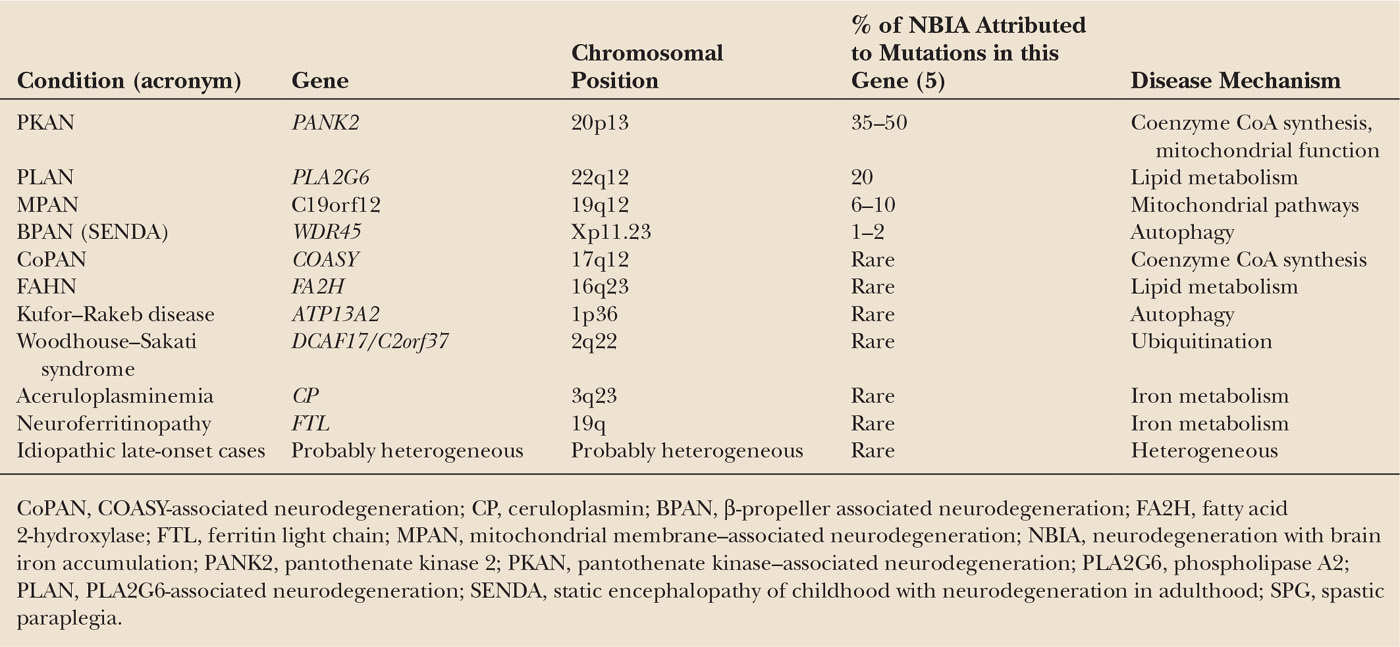
Another important milestone in the history of these diseases was the development of high-field MRI in the 1980s, which allowed noninvasive detection of iron accumulation in vivo (i.e., as hypointensity on T2-weighted images or more recently susceptibility-weighted images (SWI) etc.) (2,3). The first actual NBIA gene, PANK2, was eventually identified by Hayflick and colleagues in 2003 (4). Since then, several other genes have been identified (including PLA2G6, FA2H, C19orf12, COASY, ATP13A2, CP, and FTL) (Table 23.1). Taken together, these genes account for approximately 70% of patients with NBIA—leaving a significant fraction without an identified genetic defect (6), so more work needs to be done.
PREVALENCE OF NBIA DISORDERS
NBIA disorders are rare diseases. To remind the readers, rare diseases are those which affect less than 200,000 persons in the United States (or: affecting fewer than 5 people in 10,000 people, according to EU criteria) (prevalence rate). Data of the exact prevalence of NBIA are scarce. There have been estimates that PKAN has a prevalence of 1 to 2 per 1,000,000. However, there are local differences with some geographic areas of clustering, probably due to a founder effect like in the Caribbean, precisely in the Dominican Republic. Clustering has also been observed for neuroferritinopathy: the majority of identified cases appear to be part of an extended pedigree in the northeast of England, but unrelated patients (with different gene mutations) are increasingly being identified across the globe (7).
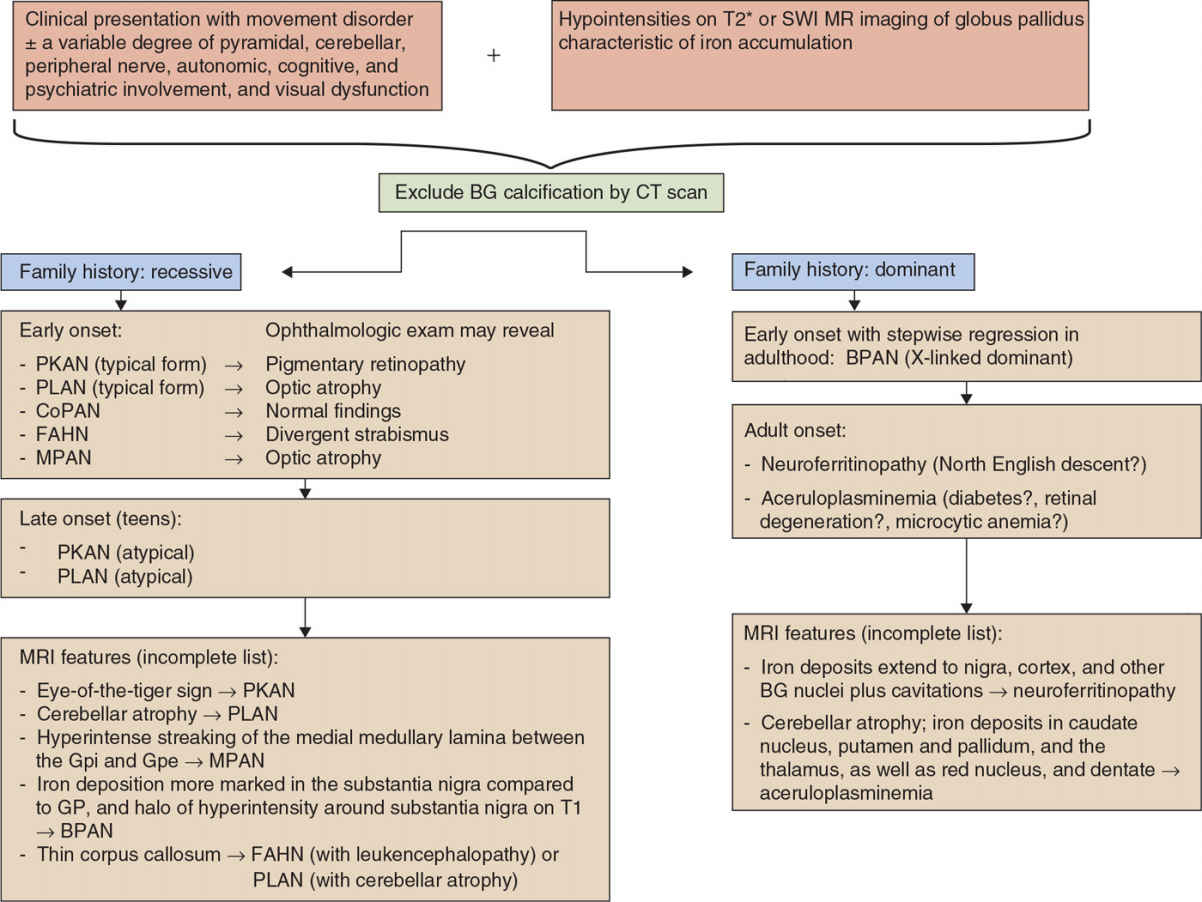
Figure 23.1. Algorithm toward the diagnosis of NBIA variants. CoPAN, COASY-associated neurodegeneration; CP, ceruloplasmin; BPAN, β-propeller associated neurodegeneration; FA2H, fatty acid 2-hydroxylase; FTL, ferritin light chain; MPAN, mitochondrial membrane–associated neurodegeneration; NBIA, neurodegeneration with brain iron accumulation; PANK2, pantothenate kinase 2; PKAN, pantothenate kinase–associated neurodegeneration; PLA2G6, phospholipase A2; PLAN, PLA2G6-associated neurodegeneration; SENDA, static encephalopathy of childhood with neurodegeneration in adulthood; SPG, spastic paraplegia; BG, basal ganglia; SWI, susceptibility-weighted images.
MODE OF INHERITANCE
Most subtypes of NBIA disorders are inherited in an autosomal recessive manner, in particular those with early onset—as a rule of thumb—including PKAN, PLAN, CoPAN, FAHN, and Kufor–Rakeb disease). In contrast, inheritance is autosomal-dominant in neuroferritinopathy and aceruloplasminemia, in which disease onset usually occurs in adulthood. Finally, de novo mutations occur in X-linked dominant NBIA (BPAN), a rare form which accounts for 1% to 2% of NBIAs (8).
NATURAL HISTORY
AGE OF ONSET
In most cases of NBIA, age of onset is in early childhood. In PLAN, first symptoms occur in infancy (infantile neuroaxonal dystrophy, INAD), although the phenotype of PLA2G6 mutations is broad and onset may occur in the late teens or early adulthood (atypical PLAN). In PKAN, onset is typically in early childhood, around age 3 to 6 years, but again, symptoms may begin later (atypical PKAN). Onset usually occurs in adulthood in two forms of NBIA, neuroferritinopathy and aceruloplasminemia, around ages 40 and 50; however, this may also vary.
DISEASE EVOLUTION
Disease progression of NBIA disorders is typically slowly progressive. In the most common form, PKAN, loss of ambulation is seen in the majority of within 10 to 15 years of diagnosis in those with a typical phenotype. Death is often related to cardiorespiratory complications (aspiration).
Of note, in BPAN disease evolution follows a stepwise progression with global developmental delay in early childhood, followed by a stable phase. In early adulthood, there is a relatively sudden onset and fast deterioration of dystonia–parkinsonism, dementia, and spasticity.
DISEASE VARIANTS
Different genetically-defined subforms have been recognized, which will be reviewed in the following.
In PKAN, as mentioned above, onset of the classic variant occurs before the age of 6 years in almost 90% (4,9), often with gait difficulty as the presenting symptom (4). The clinical picture comprises pyramidal and extrapyramidal features with prominent dystonia, often with predominant oro–lingual–mandibular and axial involvement (10). Neuropsychiatric (11,12) and visual disturbances due to pigmentary retinopathy are frequent (13). In late-onset (adult-onset; atypical) PKAN, motor involvement tends to be less severe (14), while cognitive decline and psychiatric features may be the leading, even presenting, symptoms(4,15–22).
COASY-associated neurodegeneration (CoPAN), pathophysiologically closely related to PKAN and most recently described in two individuals (6), also presents in early childhood with gait difficulty and learning disabilities. In puberty, generalized dystonia (again prominent in the oromandibular region causing dysarthria and in the legs causing dystonic paraparesis) developed. Subsequently, the clinical picture was dominated by spastic–dystonic tetraparesis, mainly of the lower limbs associated with distal areflexia in the lower limbs due to motor axonal neuropathy and a bradykinetic-rigid syndrome. Psychiatric symptoms were present. Cognitive function was normal in one but severely impaired in the other patient. Fundoscopy and further eye assessments were normal without signs of retinopathy, in contrast to PKAN and some of the other NBIA syndromes. The first cases have survival up to the third decade, probably due to residual enzyme activity. Further cases remain to be identified.
In PLAN, the second most common type of NBIA, the early-onset form presents as INAD characterized by progressive motor and mental retardation, marked truncal hypotonia, early and prominent cerebellar ataxia, pyramidal signs, and early visual disturbances due to optic atrophy (in contrast to retinopathy seen in PKAN). Late-onset PLAN has been reported: the phenotype was characterized by dystonia–parkinsonism combined with pyramidal signs, eye movement abnormalities, cognitive decline, and psychiatric features (23). Levodopa-sensitive parkinsonism was characterized by the presence of tremor, including a pill-rolling rest component, rigidity, and severe bradykinesia in line with the finding of Lewy body pathology (24–26) (see below), so the condition was subsequently assigned the PARK14 locus. Cerebellar signs and sensory abnormalities which are often prominent in the early childhood variant were absent. Notably, recently exome sequencing revealed mutations in NALCN (27) as a new cause of INAD in one family, but iron was absent. The diagnosis of INAD based on physical and neurologic examinations (however, including facial dysmorphism and skeletal anomalies not typical for PLAN), nerve biopsy studies, and brain MRI which showed cerebellar atrophy but, as mentioned, no iron (27).
The clinical picture of MPAN includes childhood-onset (mean age 10 years) dysarthria and gait difficulty due to development of spastic paraparesis with pes equinovarus foot deformities. Extrapyramidal features (dystonia and parkinsonism), motor axonal neuropathy, optic atrophy, and psychiatric symptoms and cognitive decline typically develop (28–35). However, MPAN may present quite variably, including phenotypes characterized by hereditary spastic paraplegia (36) or juvenile amyotrophic lateral sclerosis (37) (Video 23.1). Notably, late-onset (in the mid- or late 20s (38)) and mild phenotypes resembling idiopathic Parkinson’s disease (PD) have been reported.
The phenotype of FAHN is characterized by childhood-onset gait impairment, spastic quadriparesis, severe ataxia, and dystonia. Seizures and divergent strabismus may also be present. Overall, there is again remarkable phenotypic overlap with the neuroaxonal dystrophies, and the disorder is allelic to SPG35.
In BPAN (39–43), there is a global developmental delay in early childhood with neuropsychiatric symptoms, which fall into the autistic and affective spectrum (40), and the phenotype may resemble atypical Rett’s syndrome or atypical Angelman’s syndrome. However, as symptoms may be unspecific and MR imaging may be normal in early stages, diagnosis may be delayed. As mentioned above, BPAN presents with a stepwise regression: A relative sudden onset and fast deterioration of parkinsonism with variable response to dopaminergic treatment, dystonia, myoclonus, spasticity, dementia, autonomic dysfunction and epileptic seizures in adulthood, and presence of iron accumulation on brain imaging should raise the suspicion of this disorder and trigger genetic workup.
The clinical phenotype of Kufor–Rakeb disease (44–47) comprises adolescence-onset parkinsonism, with pyramidal tract signs in some. Oculogyric dystonic spasms, facial–faucial–finger mini-myoclonus and autonomic dysfunction may be present. Eye movement abnormalities with incomplete supranuclear upgaze palsy can be a clue. Psychiatric features include visual hallucinations and dementia.
Onset of neuroferritinopathy is in adulthood around age 40 years with a mixed movement disorder: The phenotype may resemble Huntington’s disease with prominent extrapyramidal features, including chorea, stereotypies, and dystonia (48) with prominent oro–lingual–mandibular dyskinesia and blepharospasm. About 10% of patients present with parkinsonism. Cerebellar ataxia is present in about half of the cases. Pyramidal involvement is usually absent. Cognitive dysfunction, depression, and psychosis may be present. Penetrance is believed to be full.
Most common presenting features of aceruloplasminemia are cognitive impairment, cerebellar ataxia, and craniofacial dyskinesia. The average age at diagnosis is around age 50 years, ranging from 16 to 71 years (49). Ophthalmologic examination usually reveals evidence of peripheral retinal degeneration secondary to iron accumulation and photoreceptor cell loss. Furthermore, all cases have evidence of systemic iron accumulation at the time of diagnosis with microcytic anemia, diabetes, and increased liver iron content (without cirrhosis). These nonneurologic symptoms frequently predate the neurologic symptoms and be a clue to the diagnosis.
MOLECULAR GENETICS AND PATHOPHYSIOLOGY
The genes associated with the various NBIA subtypes are listed in Table 23.1. Gene function remains ill understood for most of the forms, and in general no obvious genotype–phenotype correlations have yet emerged.
In PKAN, mutations, mostly missense, have been detected in all seven exons of the PANK2 gene. Deletions, duplication, and splice-site mutations as well as exon deletions also occur (50). Two common mutations account for about one-third of all PKAN cases, that is 1231G>A and 1253C>T. The majority of the remainder cases carry “private mutations” (14), including the founder mutation, 680A>G, found in the Dominican Republic. Some mutations may be associated with milder phenotypes than others (4,16).
CoA synthase (COASY), located on 17q12, is the latest of the NBIA genes. It has nine exons. The two CoPAN patients described so far were homozygous (c.1495C>T) and compound heterozygous (for the same mutation, c.1495C>T, and c.175C>T) (6).
These two forms of NBIA are linked to coenzyme A metabolism, a key molecule for the metabolism of fatty acids, carbohydrates, amino acids, and ketone bodies. PKAN is due to a defect in the first step of coenzyme A synthesis, while COASY encodes a bifunctional enzyme that catalyzes the two last steps in CoA synthesis from pantothenic acid (vitamin B5) by coupling phosphopantetheine with ATP to form dephospho-CoA and its subsequent phosphorylation to generate CoA (6). In prokaryotes, the role of COASY is carried out by two separate enzymes, phosphopantetheine adenylyltransferase (PPAT) and dephospho-CoA kinase (DPCK).
Both, PANK2 and COASY, are mainly targeted to mitochondria, and disease pathophysiology may relate to dysfunction of cellular energy metabolism. Indeed, a recent study examining blood metabolic profiles in PKAN documented elevated levels of lactate suggesting mitochondrial dysfunction and reduced levels of triglycerides, cholesterol metabolites, and sphingomyelins confirming the role of PANK2 in lipid metabolism. Cysteine accumulation as cause of iron chelation and formation of free radicals have been hypothesized (51).
Numerous different mutations are present in the PLA2G6 gene (associated with PLAN). It contains 19 exons without mutational hotspot. The encoded protein, iPLA2-β, is a group VIA calcium-independent phospholipase A2 that hydrolyzes the sn-2 acyl chain of phospholipids, thereby generating free fatty acids and lysophospholipids. iPLA2-β is thought to play a role in remodeling of membrane phospholipids, signal transduction, cell proliferation, and apoptosis. It has been suggested that in case of loss of iPLA2 function, lipid composition of the plasma membrane, vesicles, or endosomes may be altered. While no obvious genotype–phenotype has yet emerged, recent functional analysis (52) suggested that, compared to the wild-type, mutant proteins associated with INAD exhibited less than 20% of the specific activity in both lysophospholipase and phospholipase assays, which predicted accumulation of PLA2G6 phospholipid substrates. In contrast, mutations associated with dystonia–parkinsonism did not impair catalytic activity, which may explain the relatively milder phenotype and absence of iron accumulation in at least some cases.
MPAN is due to mutations in C19orf12 at chromosome 19q (53). The gene has three exons and encodes two isoforms with two alternative first exons. More than 30 different mutations have been described in the literature so far, including frameshift mutations, missense mutations, nonsense mutations, and splice-site mutations (54). Due to a founder effect in the original Polish cohort, the majority of patients described so far carry a deletion of 11 base pairs leading to a premature stop codon (Gly69ArgfsX10) and predicted to cause early truncation of the protein. Another frequent change is the p.Thr11Met variant which may be associated with later onset (54). The underlying pathophysiology of MPAN is ill understood as the function of the orphan protein is still unclear. It is localized predominantly in mitochondria, and it is coregulated with genes involved in fatty acid metabolism.
FAHN is due to mutations in FA2H located on chromosome 16q which contains 7 exons. Only a handful of cases, mostly from Asia and southern Europe (55), have been reported, including those with clinical presentations of allelic disorders (leukodystrophy and hereditary spastic paraplegia, type SPG35) (56–59). No genotype–phenotype correlations have emerged. The encoded protein catalyzes hydroxylation at position 2 of the N-acyl chain of the ceramide moiety. Glycosphingolipids, which contain a high proportion of 2-hydroxy fatty acid, are important constituents of myelin sheaths (59).
BPAN is due to mutations in WDR45 located on the X chromosome which consists of 12 exons (39–43). Initial mutations were identified by exome sequencing, confirmed by Sanger sequencing. Reported cases carried heterozygous de novo mutations, suggesting a dominant mode of inheritance. So far, no common mutation has emerged; in fact, in 20 unrelated patients 19 different mutations were identified (43). In the initial report, male and female patients had identical clinical and radiologic features and lymphoblastoid cells demonstrated exclusive expression of the mutant transcript, suggesting somatic mosaicism and X inactivation of the wild-type allele as the most likely mechanism. Little is known about the function of the gene: The encoded protein, WD repeat domain phosphoinositide-interacting protein 4 (WIPI-4), belongs to the family of WD40 proteins which promote protein–protein interactions and play a role in autophagy, cell cycle control, signal, and transduction. Patient-derived lymphoblast cell lines show decrease in autophagic flux compared to controls and accumulate early autophagic structures (39,42). WDR45 also has a β-propeller tertiary structure and a site allowing interaction with phospholipids.
Kufor–Rakeb disease is due to mutations in the ATP13A2 (60) gene located on chromosome 1p. The 26 kb-spanning gene contains 29 exons and encodes a lysosomal 5 P-type ATPase. Only a handful of cases have been reported, and there is some overlap with neuronal ceroid lipofuscinosis (NCL) (61). Most patients carried homozygous mutations, but compound heterozygous cases have also been identified (for summary, see (62)). In ATP13A2-associated neurodegeneration, lysosomal dysfunction with enhanced mislocalization of ATP13A2 to the endoplasmic reticulum may be a key mechanism (63,64). Histology revealed membrane-bound electron-dense material of lamellar appearance with inclusions in Schwann, perineural, and smooth muscle cells of the sural nerve, and skin cells, with resemblance to irregular primary lysosomes. Brain pathology of Kufor–Rakeb cases is lacking; however, the abovementioned cases diagnosed with NCL came to pathology and their brains showed abundant neuronal and glial lipofuscinosis involving cortex, basal nuclei, cerebellum, but sparing the white matter (Table 23.2). Whorled lamellar inclusions were typical of NCL in electron microscopy. Lipofuscin deposits were confirmed in the retina.
Neuroferritinopathy is caused by mutations in the FTL1 gene. The gene contains four exons, and encodes a single 175 amino acid protein, the ferritin light chain. To date, over 70 patients with at least seven pathogenic mutations have been reported, including six frameshift mutations and one missense mutation. Six of the seven mutations are located in exon 4. Insertion at position 460 accounts for most cases due to a founder effect (with clustering in Cumbria in North England), but private mutations have been observed in patients from other regions, including France, Canada, and Japan (48,66). Most mutations cause disruption of the ferritin light-chain protein, which results in an abnormal configuration of the ferritin molecule. Brain pathologic examination has revealed ferritin-positive spherical inclusions; ferritin inclusions localized extracellularly and intracellularly in iron-rich areas, often colocalizing with microglia, oligodendrocytes, and neurons. Neuroaxonal spheroids immunoreactive to ubiquitin and tau, and neurofilaments have been reported, bridging the gap to the group of neuroaxonal dystrophies discussed above. The main sites of involvement are the posterior putamen and cerebellum, but extracerebral pathology such as hepatic iron deposits may be present in some patients.
Notably, mutations in the iron-responsive element (IRE) of the FTL gene are associated with hyperferritinemia–cataract syndrome.
Aceruloplasminemia is due to ceruloplasmin gene mutations located at 3q. The gene contains 20 exons. More than 40 distinct, mostly truncating mutations leading to a premature stop codon have been reported. Most mutations are “private” mutations. No genotype–phenotype association has emerged. The encoded protein plays a crucial role in the mobilization of iron from tissues through its ferroxidase activity. Protein dysfunction results in excessive iron accumulation not only in the brain (basal ganglia, thalami, dentate nuclei, and cerebral and cerebellar cortices) but also within the retina, pancreas, and liver.
The pathophysiology of the late NBIA variants includes abnormal transport of iron (neuroferritinopathy) and copper (aceruloplasminemia) and storage of iron (neuroferritinopathy) and its mobilization from tissues. Low (neuroferritinopathy) or markedly elevated (3- to 40-fold in aceruloplasminemia) ferritin levels are diagnostic clues. Ceruloplasmin, however, is typically undetectable in the serum of homozygote patients with aceruloplasminemia.
DISEASE MECHANISMS: PATHOPHYSIOLOGY
Exact mechanisms remain ill understood, but abnormal mitochondrial function (PKAN and CoPAN), lipid metabolism (PLAN, MPAN, FAHN), and autophagy (BPAN, Kufor–Rakeb disease) have been implicated.
ANIMAL MODELS
The generation of murine and other animal models of NBIA provides clues to the underlying pathophysiology using histology and/or functional assays.
As mentioned above, two forms of NBIA are linked to the CoA biosynthetic pathway. This is in line with animal studies where abolishing different genes yielding disturbance of the fumble/PANK2 and PPAT-DPCK activities caused a neurologic phenotype characterized by brain vacuolization; albeit without iron accumulation (67). Knockout mouse models and Drosophila models of PKAN (“fumble flies” with downregulation of fumble, the Drosophila PANK homologue) demonstrate localization of the protein to mitochondria, similar to the human orthologue; and PANK2-defective neurons show an abnormal mitochondrial membrane potential (68). However, while the mice exhibit retinal degeneration and absence of sperm (azoospermia), the model do not reliably recapitulate the neurologic phenotype (68). Notably, feeding Drosophila flies pantethine led to restoration of CoA levels, improvement of mitochondrial function, and enhanced locomotor abilities (69).
Different mouse models of PLAN, both with PLA2G6 null mutations and bearing point mutations, have been developed and confirmed presence of Lewy body pathology, in addition to widespread formation of spheroids (70–73). Functional studies, amongst others, revealed severe disturbance in Ca(2+) responses to ATP in astrocytes (72).
Mouse models of FA2H-associated neurodegeneration have also recently been developed (74,75). Following a period of normal myelin development marked demyelination and profound axonal loss in the CNS occurred. Axons were abnormally enlarged, and there was abnormal cerebellar histology. In contrast, structure and function of peripheral nerves were largely unaffected.
Mouse models of Kufor–Rakeb disease have recently been reported (76). Behaviorally, Atp13a2-deficient mice displayed late-onset sensorimotor deficits (76). Increased levels of insoluble α-synuclein in the hippocampus, but not in the cortex or cerebellum, were detected; while the number of dopaminergic neurons in the substantia nigra and striatal dopamine levels were normal. Accelerated deposition of autofluorescent storage material (lipofuscin) characteristic of NCL was observed in cerebellum, hippocampus, and cortex of Atp13a2-deficient mice in line with the clinical overlap with NCL (see above). Notaby, ATP13A2 mutations have also been identified in Tibetian terriers clinically affected by NCL strengthening this link (77,78).
< div class='tao-gold-member'>





