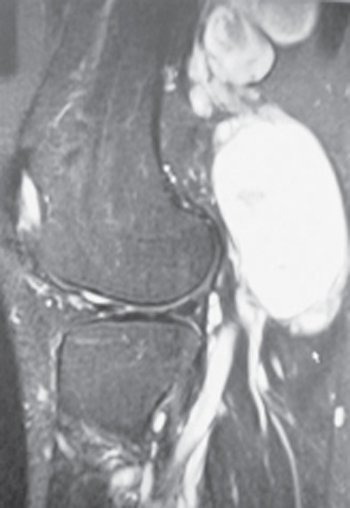
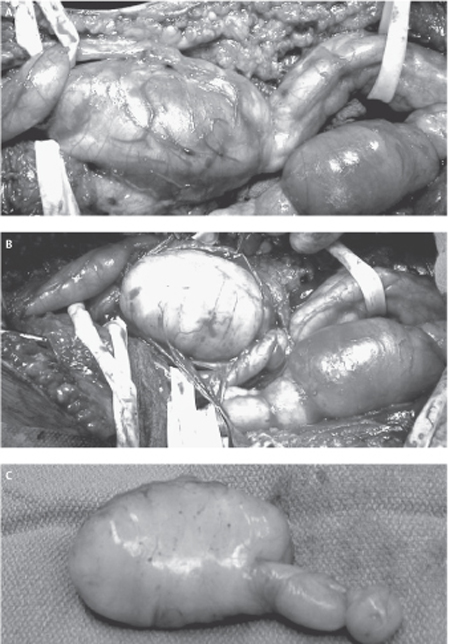
50 Neurofibroma A 34-year-old female with a known history of neurofibromatosis type 1 (NF1) presented to the neurosurgical clinic with a 4-year history of right leg pain and an increasing mass in the popliteal fossa. The patient had previously undergone surgical resection of multiple hand and facial subcutaneous neurofibromas by another surgeon. The patient described the right leg pain as dull and aching and located just behind the knee, with radiation of a sharper pain intermittently and posteriorly through the calf into the ankle. Over the past year, the pain had become more prominent and she had noted that the known lesion in the popliteal fossa was increasing in size. She has had occasional paresthesias and was aware of some numbness in the dorsum of the foot and mild weakness in her ankle movements. These neurological symptoms had been gradually progressive over 2 years. Other than having NF1, past medical history was significant for a known prolactin-secreting adenoma and hypothyroidism. The patient was pregnant, late in her second trimester. Physical examination revealed a healthy-looking, pregnant woman. Several café au lait spots were noted as were multiple small subcutaneous masses, consistent with dermal neurofibromas (Fig. 50–1). Palpation identified fullness in the posterior lower right thigh, just above the knee crease. Pressure over this region caused radiation of pain down to the ankle. Muscle power was decreased to Medical Research Council grade 4 in the right tibialis anterior and extensor hallucis longus and was barely grade 4 in plantar and toe flexion, with particular weakness noted on attempted toe walking. Sensory examination revealed decreased pinprick sensation over the dorsum of the right foot, and more profound hypoesthesia in the sural and tibial nerve distributions. The right Achilles jerk was absent. Magnetic resonance imaging revealed mass lesions involving the right sciatic nerve divisions in the popliteal region. The lesions were expanding the tibial and peroneal nerves in a plexiform fashion, and the larger tibial lesion measured 7x5x4 cm. The masses had inhomogeneous signal properties but enhanced homogeneously with gadolinium and were consistent with plexiform neurofibromas (Fig. 50–2) The lesions were well demarcated with no obvious invasion of adjacent muscle or soft tissues. Figure 50– 1 A large café au lait spot and dermal lesions are seen on the trunk of a patient with neurofibromatosis type 1. Figure 50– 2 Sagittal T1-weighted gadolinium-enhanced magnetic resonance imaging from the patient’s right knee region shows extensive lesion involving the tibial nerve. Note the nodular thickening along the nerve, characteristic of a plexiform lesion. Several months after delivery of a healthy baby, the patient underwent exploration of her right popliteal fossa. At surgery, a large plexiform lesion involving the right tibial nerve and a slightly smaller one involving the peroneal nerve were dissected out. Using microdissection techniques, aided by intraoperative electrophysiology, it was discovered that the majority of the tibial nerve lesion arose from one hugely distended fascicle, whereas two enlarged fascicles made up the majority of the peroneal lesion (Fig. 50–3A). However, all fascicles from each of these nerves were grossly abnormal but also electrically functional. It was elected to remove only the largest grossly abnormal fascicles (Fig. 50–3B,C). Histopathological examination confirmed plexiform neurofibromas, with some nuclear atypia but low mitotic activity, and proliferation index less than 1%. Postoperatively, the patient’s pain was completely alleviated but she was initially slightly worse in motor function involving both the tibial and the peroneal nerve distribution, yet still much better than antigravity function. Over longer-term follow-up, she has regained motor function to the same level as her preoperative examination. Figure 50–3 (A) All three divisions of the sciatic nerve are abnormal with most if not all fascicles enlarged by intraneural tumors. (B) Following electrical mapping of the tumor surface, the dominant fascicle in the tibial nerve involved by tumor is dissected and removed. (C) No attempt is made to remove the other less involved fascicles to preserve postoperative neurological function. Neurofibroma (of sciatic nerve divisions) Before discussing the typical clinical presentation of neurofibromas, two different lesion types need to be delineated. The first is the fusiform neurofibroma, which usually arises from a single nerve fascicle as an isolated proliferation of neoplastic cells within the sheath of an associated nerve. It develops as a nodular swelling on a nerve. These may be solitary lesions, but are often multiple and occur as part of neurofibromatosis type 1 (NF1). Plexiform neurofibromas constitute the second type of neurofibroma. These tumors form a network-like growth, arise from multiple nerve fascicles, often involve multiple branches of a large nerve, and lead to a diffuse mass of thickened nerves. Plexiform neurofibromas do not necessarily involve a nerve plexus such as the brachial or lumbosacral plexi. Plexiform neurofibromas are almost exclusively associated with NF1. As alluded to earlier, neurofibromas may occur as isolated lesions or in association with NF1. NF1 is associated with different subtypes of neurofibromas, and the clinical implications of these tumors with respect to surgical management and follow-up are different. Given these issues, a discussion of the clinical presentations of neurofibromas is best done within the context of those occurring in relation to NF1 and those not associated with NF1. Neurofibromas that occur outside of the NF1 syndrome are almost exclusively solitary, fusiform lesions. Like schwannomas, they may occur within any nerve distal to the oligodendroglia-Schwann cell interface, and thus are found in association with nerves ranging from small unidentifiable cutaneous nerves, larger peripheral nerves, spinal nerve roots, or the brachial and lumbosacral plexi. Unlike schwannomas, neurofibromas of cranial nerves are exceedingly rare. Although schwannomas display a greater tendency to arise from sensory portions of nerves, neurofibromas exhibit a predilection for the motor portion. In general, these lesions are more likely to occur in females than in males. They preferentially occur on the right side of the body, the reason for which is unclear, and it may simply be an epidemiological artifact. As with schwannomas, a painless, palpable mass is the most common presentation. However, although pain as a presentation occurs in a minority of patients (38% in one large clinical series), neurofibromas are more likely to be painful lesions than other nerve sheath tumors. This pain is almost exclusively radicular in nature. Neurofibromas are laterally, but not longitudinally, mobile, along with their nerve of origin. A positive Tinel sign is quite common. Patients harboring neurofibromas may also complain of sensory loss, paresthesias, or weakness in the distribution of the involved nerve. Such objective loss of function occurs in the minority of patients but occurs more commonly with neurofibromas than with schwannomas. The more infiltrative nature of the neurofibromas, as will be discussed later, accounts for this observation. The potential for malignant degeneration of neurofibromas that occur outside the context of NF1 is extremely low, and akin to that of schwannomas. As with schwannomas, new onset or greatly increased pain, rapid decreased function in the distribution of the nerve, and rapid tumor growth are all signs that should raise suspicion of malignant degeneration. NF1 is one of the most common autosomal dominant disorders in humans and is the commonest cancer-causing inheritable disorder. In addition to its heritable transmission, fully 50% of NF1 cases represent new mutations. Population-based studies suggest that the prevalence of NF1 approaches 1/4000, whereas the incidence among new births is 1/2500. The NF1 gene is located on the long arm of chromosome 17 and spans over 350 000 base pairs. It is the extremely large size of the gene that likely accounts for the high incidence of mutation. The NF1 gene codes for the protein neurofibromin, a putative tumor suppressor.
 Case Presentation
Case Presentation
 Diagnosis
Diagnosis
 Characteristic Clinical Presentation
Characteristic Clinical Presentation
Neurofibromas Not Associated with NF1
Neurofibromatosis Type 1– and 2– Associated Neurofibromas
| Two or more of the following: |
| I.6 café au lait macules measuring ≥ 5 mm in prepubertal individuals and ≥ 15 mm in postpubertal individuals |
| II.≥ 2 neurofibromas of any type |
| III. ≥ 1 plexiform neurofibroma |
| IV. Axillary and/or inguinal freckling |
| V. Optic glioma |
| VI. ≥ 2 Lisch nodules (i.e., benign iris hamartomas) |
| VII. A distinctive bony lesion (e.g., sphenoid wing dysplasia, cortical thinning of a long bone) |
| VIII. A first-degree relative with neurofibromatosis type 1 |
The NF1 gene exhibits full penetrance and variable expression, demonstrating a wide range of clinical features that can involve practically any organ system. Diagnostic criteria for NF1 have been established by a National Institutes of Health (NIH) Consensus Conference and are detailed in Table 50–1. In addition to the major defining features outlined in Table 50–1, there are many minor features and associated abnormalities that may be found among the NF1 population. These include macrocephaly, short stature, neurodevelopmental delay, and seizure disorders. A detailed account of the wide-ranging spectrum of NF1 and the clinical management of NF1 sufferers is beyond the scope of this Chapter, and the reader is referred to selected readings in the reference section.
In addition to the “classic” NF1 phenotype, a somatic mosaic variant exists that results in signs of NF1 limited to one or more body parts. This has been termed segmental neurofibromatosis, or NF5, and has an estimated frequency of 1/70,000 to 80,000. Affected individuals may have pigmentary abnormalities, multiple fusiform neurofibromas, plexiform neurofibromas, or a combination of these features isolated to a single body region.
Neurofibromas are the most consistent feature of NF1, and as stated earlier, both fusiform and plexiform varieties occur in association with NF1. Practically all NF1 patients will develop cutaneous, or dermal, neurofibromas by the time they reach adulthood. These lesions are fusiform neurofibromas of tiny cutaneous nerves. They are soft, discrete nodules, often with a violaceous color, which lie with the dermis and epidermis. They can occur on any cutaneous surface, are rarely painful, and exhibit a nonlinear growth pattern with years of slow growth followed by periods of rapid expansion. These lesions only extremely rarely undergo malignant degeneration, but unfortunately, due to their usual occurrence in large numbers, are often severely disfiguring.
NF1-associated fusiform neurofibromas have a peak incidence in the late third decade, occurring somewhat earlier than solitary neurofibromas, whose peak incidence is ˜10 years later. No male/female predilection exists for NF1-associated neurofibromas. Like other neurofibromas these lesions may present as a palpable, painless mass. However, loss of function (motor and/or sensory) in the distribution of the involved nerve appears more common among NF1-associated neurofibromas, with only ˜20% of cases exhibiting normal preoperative function.
Plexiform neurofibromas are common in NF1 patients, occurring almost exclusively in relation to NF1. One study of computed tomographic (CT) screening of NF1 patients documented plexiform neurofibromas in 44% of patients, most of which were clinically asymptomatic. Other studies suggest that ˜25% of NF1 patients have plexiform neurofibromas evident on clinical examination. They generally present as a large, soft, subcutaneous swelling with poorly defined margins in early childhood, or at the latest, in early adulthood. Often the overlying skin is hyperpigmented or hypertrophied or both. Extreme clinical cases are termed elephantiasis neuromatosa. Occasionally, excessive hypertrichosis is associated. Although severe intractable pain may be associated with plexiform neurofibromas, this is usually the exception rather than the norm. Motor and sensory abnormalities along the affected nerve are very common.
The risk of sarcomatous degeneration is quite high among nondermal NF1-associated neurofibromas and is a particularly worrisome feature of plexiform neurofibromas. In general, NF1 patients have a 5% incidence of malignant peripheral nerve sheath tumors (MPNSTs), which represents a relative risk of 4000 over the general population. Malignant degeneration is thought to occur in ˜15% of NF1-associated neurofibromas. Some case reports also suggest that the risk of malignant degeneration in neurofibromas of patients with segmental neurofibromatosis may be intermediate between the general population and NF1 sufferers. As such, neurofibromas need to be carefully followed in all neurofibromatosis patients, and any hint of symptomatology that could suggest malignant progression should trigger aggressive surgical therapy.
 Differential Diagnosis
Differential Diagnosis
Whenever a patient with a known history of neurofibromatosis presents with a nodular swelling of an extremity the possibility of a peripheral nerve sheath tumor should top a list of possible diagnoses. Of course, one cannot neglect to entertain the general differential diagnoses pertaining to a nodular swelling of an extremity, as was discussed in Chapter 49 on schwannomas. Table 49–1 from that Chapter also provides a general framework with which to approach peripheral nerve tumors. From this list, neurofibromas, schwannomas, and MPNSTs are the most likely possibilities in a patient with NF1. In almost all situations, clinical signs and symptoms, as well as appropriate diagnostic imaging, will help to delineate the first two from the latter, but this is not always possible. When considering neurofibroma in the differential diagnosis, especially against the backdrop of NF1, it is important to separate fusiform and plexiform neurofibromas because the goals of management are significantly different, as will be detailed in the following sections. Again, clinical history and examination as well as imaging will help to delineate these entities.
 Diagnostic Tests
Diagnostic Tests
When investigating a nodular swelling of an extremity, diagnostic imaging modalities such as ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI) are very important. Such studies help to delineate the anatomy of the region under investigation and define the relation of the lesion to nerve structures. The possibility of the lesion in question being a peripheral nerve neoplasm hence becomes readily apparent. However, as stated in Chapter 49 on schwannomas, it is impossible to definitively delineate a neurofibroma from a schwannoma through imaging characteristics alone because their imaging features greatly overlap. As discussed in the previous Chapter, ultrasound can be used to quickly image peripheral nerve tumors, where a well-defined mass of variable acoustic enhancement is suggestive of a peripheral nerve tumor. However, the spatial resolution is not sufficient to delineate surrounding structures adequately and, as such, is of little use in preoperative planning. The CT appearance of neurofibromas is similar to schwannomas, demonstrating a hypodense soft tissue mass that is homogeneous both before and after contrast administration. MRI offers superior anatomical detail, often demonstrating a nerve of origin, and is the imaging modality of choice. Like schwannomas, neurofibromas generally are isointense on T1-weighted sequences and bright on T2-weighted images. Neurofibromas are homogeneously enhancing. Plexiform neurofibromas can often be demonstrated on CT and MRI to consist of a tortuous, thickened mass of nerves with sequence appearances as already outlined.
As stated with respect to schwannomas, nerve conduction studies and electromyography (NCSs/EMG) do not contribute specifically to the diagnosis of a neurofibroma. However, because patients with neurofibromas, especially plexiform types, often complain of functional impairment in the distribution of the affected nerve, NCS/EMG can offer objective measurement of nerve function prior to treatment initiation and provide an objective measure of treatment outcome. As such, they remain an important preoperative investigation.
 Management Options
Management Options
Historically, the treatment of neurofibromas has been an area of great controversy. Many surgeons had previously advocated that neurofibromas could not be totally resected without a significant risk of damaging the parent nerve. This view undoubtedly arose from the traditional teaching that neurofibromas are unencapsulated tumors. Within this same line of reasoning, others had advocated en bloc excision with end-to-end reanastomosis or sural nerve graft repair of the parent nerve as the only method to obtain gross total resection. However, in recent years numerous surgical series have made it evident that these historical teachings were, for the most part, erroneous, and indeed most neurofibromas are encapsulated. With proper surgical technique, a gross total excision can be achieved of the fusiform lesions without undue risk to neurological function.
Currently, as with schwannomas, surgical resection is the mainstay of therapy for neurofibromas. Practically all of the surgical principles for schwannoma resection also hold true for neurofibromas, especially those tumors occurring in patients who do not have NF1. However, due to the more infiltrative nature of neurofibromas, with encasement of numerous nerve fascicles, surgical technique must be particularly meticulous to avoid deterioration of neurological function postoperatively (Fig. 50–3B).
Non-NF1 fusiform neurofibromas have an extremely low malignant potential, on par with that of schwannomas. Surgical indications are similar to schwannomas and include refractory pain (radicular and/or local), progressive neurological deficit, and local compressive symptomatology, as well as patient preference and cosmesis.
Due to the unencapsulated and extremely infiltrative nature of plexiform neurofibromas, surgical excision is often incompatible with functional preservation. Of course, these lesions also have a high rate of sarcomatous transformation and must be followed closely. Patients with severe pain or other signs of potential malignant degeneration should be offered surgery, with the expectation that there will be a postoperative functional loss.
As already stated, the basic surgical principles used for schwannomas also apply to neurofibromas. This includes wide surgical prepping and draping, such that the distal musculature of the involved nerve is visible and accessible, as well as an anesthetic protocol without neuromuscular blockade. This allows for intraoperative EMG and nerve action potential (NAP) recording of the involved nerve and distal musculature. Incisions should adequately expose the involved nerve beyond the proximal and distal poles of the tumor and allow for prophylactic release of any entrapment points near the resection bed.
Once the neurofibroma is exposed it is usually quite easy to identify numerous fascicles adherent to the capsule, as well as some fascicles lying within its layers. Using microsurgical techniques, one should proceed with dissection of all adherent fascicles from the periphery of the lesion, as well as those fascicles within the capsule, leaving the capsule attached to the tumor. The maneuver is best facilitated by addressing the proximal pole of the tumor first, identifying the fascicle, or fascicles, that give rise to the neurofibroma, and dissecting out the remaining fascicles toward the tumor. In the same manner the distal pole can then be addressed, again carefully dissecting out fascicles toward, and not away from, the tumor. NAP studies are then performed on all fascicles that enter and leave the neurofibroma. Usually, the NAP tracings are flat, suggesting that these fascicles are nonfunctioning and can be sacrificed with impunity. If, however, NAPs are positive for select fascicles, these should be traced through the lesion and spared as much as possible. Such a microsurgical extracapsular excision can usually be accomplished in smaller fusiform neurofibromas. However, in larger fusiform neurofibromas, opening of the capsule and evacuation of the contents of the tumor are required to safely free adherent fascicles. An ultrasonic aspiration device is ideal for this maneuver. Rarely, functioning nerve fascicles have to be resected to obtain adequate tumor excision. Sural nerve cable grafts can be used to bridge the resulting defect. In certain situations, particularly among NF1 patients with plexiform, large fusiform, or multiple smaller neurofibromas along the course of the involved nerve, the efficacy of grafting procedures is quite low.
The infiltrative, unencapsulated growth pattern of plexiform neurofibromas precludes functional preservation in the face of total excision. Radical resections of plexiform lesions along with the parent nerve, followed by grafting procedures have also met with little success. Subtotal excision is possible, but residual tumor can regrow. As such, operative intervention for plexiform neurofibromas should be limited to palliative procedures for substantial debulking in the setting of intractable pain or progressive neurological deficit, or to rule out sarcomatous changes in a highly suspicious lesion.
 Outcome and Prognosis
Outcome and Prognosis
Total excision of solitary fusiform neurofibromas approaches 80% in the largest surgical series, and ranges from 65 to 70% for neurofibromas associated with NF1 in various series. The discrepancy is attributed to the higher incidence of plexiform lesions in the latter group. In the largest surgical series to date, published by Kline and colleagues, among the non-NF1 neurofibroma population, functional preservation was possible in 80%, whereas of those with some degree of preoperative impairment two thirds improved and only 10% further deteriorated. Of those with preoperative pain, only 14% were unchanged or worse with respect to their pain symptoms postoperatively. In the same series, among NF1-associated neurofibromas, the rates of functional preservation were comparable to non-NF1 lesions. Of those with preoperative functional impairment 50% improved and 17% were worse. Only one fourth of those with preoperative pain did not benefit from the procedure.
Unfortunately, the results for plexiform neurofibromas are uniformly poor, with all patients experiencing deterioration of neurological function. In all patients who did not undergo excision of the involved nerve and subsequent graft repair, the tumor recurred in less than 2 years.
These results highlight that with proper microsurgical techniques and intraoperative electrophysiological investigations, an experienced surgeon can safely and effectively resect fusiform neurofibromas. These results are a benchmark against which other operative results should be compared.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


