Neurogenetics
Patricia E. Greenstein
▪ INTRODUCTION
This chapter will outline the importance of history taking, and pedigree construction and analysis when suspecting a genetic component to a neurological disease. It will emphasize some of the genetic components of neurological diseases important to neurologists and psychiatrists. Genetic diseases cover a vast area that exceeds the scope of this chapter. Neuromuscular disorders have been covered in other chapters.
Genetics plays a very important role in the pathophysiology of human disease. In neurological disorders, it is widely accepted that mendelian inheritance can account for 5% to 10% of these diseases. Mendelian traits represent the most basic and simple patterns of inheritance. Nonmendelian traits represent some complexity in their mode of inheritance, in which the classic pattern may not always apply and epigenetic factors are associated with disease mechanisms. Regardless of the mode of inheritance, defining specific genetic factors that are associated with certain neurological diseases and their functional role are very important in patient management and genetic counseling, as well as understanding the mechanism of the disease to ultimately develop new therapeutics.
It is estimated that there are approximately 20,000 to 30,000 genes in the human genome. Approximately one third of these genes are expressed in the nervous system, and thus a phenotype with neurological features is common.
▪ TAKING A GENETIC HISTORY
Looking for neurological disease genes in a patient or the family begins with a clear description of the phenotype. This is the observable expression of the genetic information (genotype) as a structural, clinical, and cellular or biochemical trait.
What then follows is a detailed family history. When taking the history, it is useful to draw a detailed pedigree of the first-degree relatives (e.g., parents, siblings, and children), because they share 50% of genes with the patient.
The family history should include information about ethnic background, age, health status, consanguinity, and (infant) deaths. Next, the physician should explore whether there is a family history or illnesses related to the current problem. Because of the possibility of age-dependent expressivity and penetrance, the family history will need intermittent updating. If the findings suggest a genetic disorder, the clinician will have to assess whether some of the patient’s relatives may be at risk for carrying or transmitting the disease. In this circumstance, it is useful to confirm and extend the pedigree based on input from several family members. This information may form the basis for carrier detection, genetic counseling, early intervention, and prevention of a disease in relatives of the index patient.
The patterns shown by single-gene disorders in pedigrees depends on two factors: the chromosomal location of the gene locus, which may be autosomal (located on an autosome or nonsex chromosome), and whether the phenotype is dominant (expressed when only one
chromosome of a pair carries the mutant allele, despite the normal allele expressed on the other chromosome of the pair) or recessive (expressed only when both chromosomes of a pair carry a mutant allele).
chromosome of a pair carries the mutant allele, despite the normal allele expressed on the other chromosome of the pair) or recessive (expressed only when both chromosomes of a pair carry a mutant allele).
TABLE 29.1 PATTERNS OF MENDELIAN INHERITANCE | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Thus there are four basic patterns of mendelian inheritance (Table 29.1).
▪ MENDELIAN INHERITANCE
Autosomal Dominant Inheritance
A phenotype expressed in the same way in both homozygotes and heterozygotes is dominant, that is, if only one of the two alleles is mutated. Males and females are equally affected. Looking at a pedigree, there is male-male transmission. There is a 50% risk to each child of an affected parent. Multiple generations are affected, and expression of the phenotype can be highly variable (Fig. 29.1).
Some examples of diseases with autosomal dominant inheritance include Huntington’s disease, spinocerebellar ataxia, myotonic dystrophy, and inherited forms of Alzheimer’s disease.
Autosomal Recessive Inheritance
Autosomal recessive disease occurs only in homozygotes, individuals with two mutant alleles and no normal alleles. In these diseases, one normal gene copy in a heterozygote is able to compensate for the mutant allele and prevent the disease from occurring. A typical pedigree is shown in Figure 29.2.
In this typical pedigree, in which males and females are equally affected, there is a 25% risk of homozygosity to children of carrier parents. There is a single generation affected, and consanguinity is sometimes present. Consanguineous marriages occur in people related by descent from a common ancestor. This increases the chance that both parents are carriers of a mutant allele at the same locus.
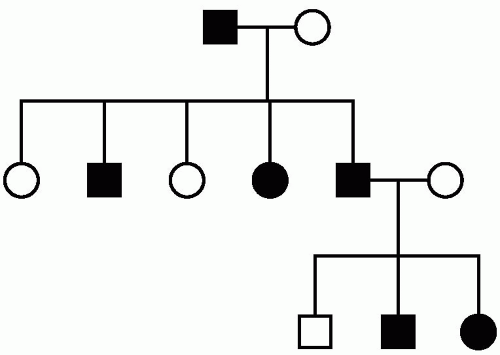 ▪ FIGURE 29.1 Autosomal dominant inheritance. |
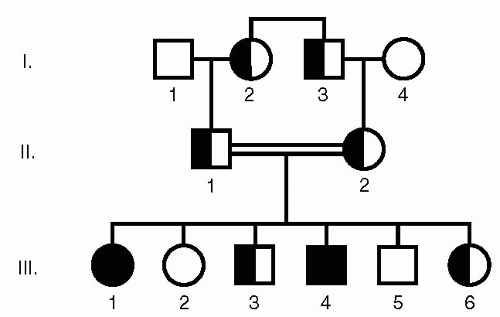 ▪ FIGURE 29.2 Autosomal recessive inheritance. |
Some examples of neurological diseases that are inherited in an autosomal recessive fashion are Friedreich’s ataxia, Tay Sach’s disease, and Canavan’s disease.
X-Linked Recessive Inheritance
The inheritance of X-linked recessive phenotypes follows a defined pattern and is typically expressed in all males who receive it, but only in those females who are homozygous for the mutation. Thus X-linked recessive diseases are generally restricted to males, with the exception of rare manifesting female carriers, who are generally more mildly affected, as determined by the pattern of X-inactivation. The gene responsible for the condition is transmitted from an affected man through all of his daughters. The gene is ordinarily never transmitted directly from father to son. Isolated cases are usually the result of a new mutation.
A typical pedigree is illustrated in Figure 29.3. Examples of such diseases are Duchenne’s and Becker’s muscular dystrophy.
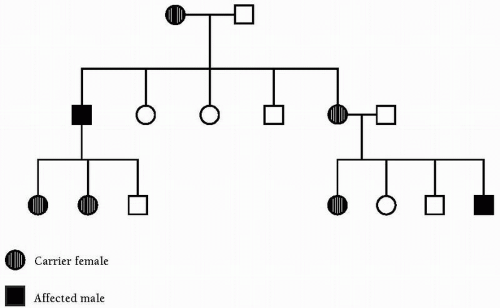 ▪ FIGURE 29.3 X-linked recessive inheritance. |
 ▪ FIGURE 29.4 Mitochondrial inheritance. |
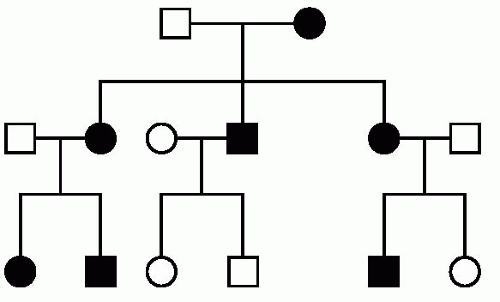
X-Linked Dominant Inheritance
As noted previously, an X-linked phenotype is described as dominant if it is regularly expressed in heterozygotes. The distinguishing feature of a fully penetrant X-linked dominant pedigree is that all the daughters and none of the sons of affected males are affected. If any daughter is unaffected or any son is affected, the inheritance must be autosomal and not x-linked.
Examples of this that are important to neurologists and psychiatrists are Rett’s syndrome, incontinentia pigmenti, and fragile X syndrome.
▪ MITOCHONDRIAL PATTERNS OF INHERITANCE
The mitochondrial chromosome is an approximately 16.5-kb circular DNA molecule (mtDNA) located outside the nucleus of the cell in the mitochondrial organelle (Fig. 29.4). The ovum and not the sperm supplies the zygote with all of its mitochondria. Therefore a mother carrying a mutation in mtDNA will pass the mutation on to all of her offspring, whereas a father carrying the mutation passes it to none. Defects in mtDNA demonstrate maternal inheritance. The other unique feature of mitochondrial inheritance is that there is passive segregation of mitochondria at cell division. In other words, the mtDNA replicates and sorts randomly among newly synthesized mitochondria, which are then distributed between the two daughter cells. This results in variation in proportion of mutant and wild-type mitochondria, a concept known as “heteroplasmy.” Mitochondrial function is essential to nearly all cells, and the phenotypic expression of a mutation in mtDNA depends on the relative proportions of normal and mutant mtDNA making up different tissues. This balance between “wild-type” and “mutant” mtDNA accounts for reduced penetrance, pleiotropy, and variable expression within a pedigree.
KEY POINTS (MITOCHONDRIAL INHERITANCE)
1. Transmission is along the maternal germline.
2. All children are at risk.
3. None of an affected male’s children are affected.
4. Highly variable expression and severity.
5. Cytoplasmic inheritance.
6. Heteroplasmy (mixing of wild-type and mutant mtDNA).
▪ POLYGENIC INHERITANCE
Some syndromes are presumed to be the result of the additive effect of a small number of multiple genes. This is still speculative, but could include some forms of mental retardation, hypertension, epilepsy, or nonmendelian dementias.
▪ MULTIFACTORIAL INHERITANCE
Multifactorial inheritance refers to disorders that result from the combination of an inherited genetic disposition acting in concert with an environmental insult, for example, acute intermittent porphyria following phenobarbital administration.
This concept has clinical relevance. It is important in the management of patients with seizure disorders who are fast or slow metabolizers. Another example is multiple sclerosis, in which there is a presumed immunological predisposition to some unknown environmental trigger. Most common neurodevelopmental disorders have complex inheritance, as do autism and most dementias. The key aspects to understanding complex genetic traits include the following:
Many genes contribute to the phenotype.
Each gene contributes only a portion to the genetic risk.
Variants commonly present in the general population may contribute.
Environmental influences may also contribute.
The disease status itself may be an arbitrary cutoff of what is a continuous distribution of a phenotype.
▪ CHROMOSOMAL DISORDERS
Abnormalities of chromosomes may be either numerical or structural and may involve one or more autosomes, sex chromosomes, or both. The most common type of chromosomal abnormality is aneuploidy, an abnormal chromosomal number resulting from an extra or missing chromosome.
Clinical phenotypic features that are important to recognize when considering a chromosomal disorder include the following:
Multiple birth defects
Involvement of midline structures: brain, heart, kidneys, gastrointestinal system
Bilateral inguinal hernias
Mental retardation
Dysmorphic facial features
The chromosomal disorders that are important for neurologists and psychiatrists to recognize include trisomies 21 (Down’s syndrome), 13, 18, and 47,XXY (Klinefelter’s syndrome).
▪ ATYPICAL PATTERNS OF INHERITANCE
Exceptions to Mendelian inheritance do occur in unusual single-gene disorders and must therefore be considered when evaluating a patient.
The two disorders relevant to neurologists and psychiatrists are Prader-Willi syndrome (PWS) and Angelman syndrome (AS). They are two contiguous-gene deletion syndromes in which multiple genes are deleted from a specific region of chromosome 15q11-q13. The diagnosis can be made on a high-resolution karyotype (analysis of the chromosomes), but more definitively by fluorescence in situ hybridization (FISH) analysis of the Prader-Willi critical region. In this analysis a fluorescent probe is hybridized to some genomic DNA from 15q11-q13. The hybridized probe fluoresces when the chromosomes are viewed with a wavelength of light that excites the fluorescent dye. If there is a deletion on one of the chromosomes, it will be visible under a microscope.
PWS is a relatively common dysmorphic syndrome with the following phenotypic features: severe hypotonia at birth, obesity, short stature, hypogonadism, and mental retardation. In 70% of cases, there is a detectable cytogenetic deletion on the long arm of chromosome 15 inherited from the patient’s father. Thus the genomes of these patients have genetic information in 15q11-q13 derived entirely from their mothers. By contrast, in AS there is a deletion of approximately the same region, but on the chromosome 15 inherited from their mother. Patients therefore have their entire 15q11-q13 information derived only from their fathers. The phenotype of these patients is also very different and includes short stature, severe mental retardation, seizures, and spasticity (“the happy puppet”).
KEY POINTS
1. Diagnosis of a contiguous-gene deletion syndrome is usually by karyotype analysis and FISH.
2. These disorders have well-described phenotypes.
3. Multiple genes are deleted, and usually patients have mental retardation.
▪ GENETIC COUNSELING AND ETHICAL ISSUES
Genetic counseling is a process by which a trained medical professional explains the genetic contribution of a disorder to a patient. When a disorder is suspected of being heritable, however, there is an added dimension—the need to inform other family members of their risk and the means available to them to modify their risk.
Just as the unique feature of genetic disease is its tendency to recur within families, the unique aspect of genetic counseling is its focus, not only on the original patient (proband) but also on the patient’s family, both present and future.
The medical professionals trained to provide genetic counseling include physicians, those who have doctorate-level training in medical genetics, certified genetics counselors, and nurses with special training in medical genetics.
During the process of genetic counseling, patients are not told what decisions to make with regard to the various testing and management options, but, instead, are provided information and support. This approach to counseling, referred to as nondirective counseling, has been adopted widely as the standard of practice in the field. Genetic counseling is not limited to giving information and calculating risk for disease, but it is rather a communication process that requires understanding of the complex psychosocial issues associated with a genetic disorder in a family.
There are many potential pitfalls in interpreting genetic test results. Most tests have a high degree of reliability, but there are opportunities for misinterpretation. These are most specifically related to linkage-based indirect tests or detection of benign polymorphisms (variants) in direct testing. Patients found to carry specific mutations may be perceived as “different” by themselves, members of their families, or members of society. There can be a great deal of guilt and altered self-image and self-blame, particularly if the gene they carry is transmitted to a child or other members of their family.
Unlike many medical tests, genetic test results may have implications for many members of a family. At all times the privacy of individuals within the family must be respected and the risks of discovery of information about others in the family discussed before testing.
This particularly applies to presymptomatic counseling of family members at risk for diseases, in particular, Huntington’s disease (HD). The counseling is particularly complex given that HD is a progressive degenerative disease for which there is no treatment or cure. Fewer than 10% of at-risk people seek testing, and concerns have been raised that predictive testing may lead to an increase in deaths by suicide among identified carriers. However, there is evidence to suggest from looking at large centers who offer presymptomatic testing that people who choose to be tested are psychologically selected for a favorable response to testing. It is recommended, however, that
presymptomatic counseling be offered only in established centers with good psychiatric or psychological support services. People with high levels of depression or hopelessness should be referred for psychiatric counseling before decision making about genetic testing.
presymptomatic counseling be offered only in established centers with good psychiatric or psychological support services. People with high levels of depression or hopelessness should be referred for psychiatric counseling before decision making about genetic testing.
▪ GENETICS OF ALZHEIMER’S DISEASE AND DEMENTIA
Genetic factors play an important role in many types of dementia. The most common form of dementia is Alzheimer’s disease (AD). AD is frequently divided into early onset and late onset, with the dividing line being 60 to 65 years of age.
Three genes are known to cause early-onset AD. The first is the amyloid precursor protein or APP gene. APP is normally cleaved at the beta and gamma secretase sites to produce a 40-42 amino acid, the beta-peptide. It is this peptide that is deposited in excess in the brains of persons with AD. The most common mutation in this gene is at position 717 and leads to the increased production of the Abeta peptide. The importance of this peptide in AD pathogenesis is further highlighted by the associated plaques and tangles in people with Down’s syndrome (trisomy 21) with three copies of the APP gene.
Mutations in presenilin-1, a gene on chromosome 14, are also a rare cause of autosomal dominant, young-onset AD. The families with mutations in APP and PS-1 typically have onset in their 40s and live for 6 to 10 years.
Presenilin-2 is a homolog of PS-1 on chromosome 1. Mutations in PS-2 also cause early-onset autosomal dominant AD in rare families. There is a wider range of age of onset from 40 to 75 years. Of these three genes, PS-1 is by far the most common. Even so, all mutations in these three genes represent less than 2% of all AD.
Late-onset AD is by far the most common type of dementia. It is generally accepted that there must be several genetic factors playing a role. The only factor recognized and confirmed is Apo E. Apo E is an important lipid and cholesterol transporter. There are 3 alleles: E2, E3, and E4. One form is inherited from each parent. The E4 allele is a significant AD risk factor. The role for APOE as a major susceptibility locus in AD was suggested by four independent lines of evidence: linkage analyses in late-onset families with an aggregation of AD, increased association of the E4 allele with AD patients compared with controls, the discovery that the Apo E protein is a component of the AD amyloid plaque, and the finding that Apo E binds to the Abeta peptide. Normal function in neurons is not known, but, as mentioned, is a component of an amyloid plaque. That being said, it is neither necessary nor sufficient for the disease.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


