Figure 86.1. Pathophysiology of neurogenic pulmonary edema.
86.2.3 Diagnosis
There are no pathognomic signs which allow the identification of NPE. Its semiology is similar to all the other cases of acute pulmonary edema (dyspnea, hypoxemia, crackles on pulmonary auscultation), but signs of left ventricular failure are frequently absent (absence of cardiac gallop).
Chest films show bilateral pulmonary opacities, corresponding to an increase of interstitial and alveolar liquid (Figure 86.2). Pulmonary infiltrates can resolve rapidly with adequate therapeutic management. In the absence of coexisting cardiac injury, the electrocardiogram does not show major changes and central venous pressure is normal. Although it would be expected to find highly elevated pulmonary capillary wedge pressure, it is not so uncommon to find those pressures within relatively normal range [5]. This is probably due to the fact that the increase in pulmonary wedge pressure is severe but temporary. As a consequence, the pressure can be normalized by the moment the pulmonary hemodynamic measures become available.
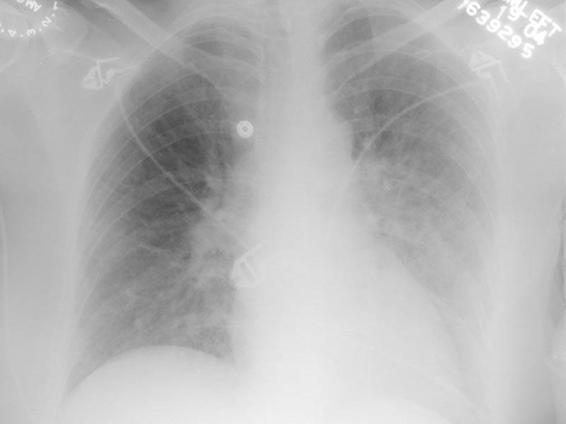
Figure 86.2. Chest X-ray findings in a case of neurogenic pulmonary edema.
The differential diagnosis should include, as in all the patients with acute hypoxemia, ventilator-associated pneumonia, aspiration pneumonitis, atelectasis, acute pulmonary embolism and cardiogenic edema from acute cardiac failure [2,4].
Furthermore it is useful to keep in mind that some hypoxic patients during the acute phase of aSAH can show relatively normal findings on chest X-ray, but this does not exclude the possibility of a of ventilation-perfusion mismatch caused by an increase of intrapulmonary liquid [5]. Once that the other causes of hypoxemia have been excluded, it is appropriate to consider these cases of cryptogenic hypoxemia as possible less severe cases of NPE.
86.2.4 Treatment
The treatment of NPE is based on the control of the acute neurological disease, with special attention to the reduction of intracranial pressure, and on providing ventilation support to improve hypoxemia [1].
Even if pulmonary complication can be fatal, the majority of deaths in these cases occur as a consequence of the primary cerebral injury. Serial assessment of neurological status and, when indicated, continuous monitoring of intracranial pressure and cerebral perfusion pressure are essential to treat these patients effectively.
Temporary endotracheal intubation and sedation are usually recommended. Some less severe cases can be managed with non-invasive ventilation, but the high levels of positive end-expiratory pressure (PEEP) necessary to treat the hypoxemia may not be well tolerated when are administered via BiPAP mask. Usually PEEP values between 10 and 15 mmHg should be used in cases of severe or moderate NPE. In general, these levels do not compromise cerebral perfusion [10], but the effect of PEEP in the intracranial and cerebral perfusion pressures should be evaluated in each single case. Hyperventilation can be necessary to achieve a fast control of the intracranial hypertension, but prolonged hyperventilation and hypocapnia (PCO2 <30 mmHg) should be avoided because the resulting cerebral vasoconstriction can cause ischemia. On the other side, permissive hypercapnia, which can be considered an adequate ventilation technique in other cases, is not indicated in the majority of patients with NPE because it can result in worsening of the intracranial hypertension. Ventilation in prone position can be a particularly advantageous option in some patients, and in my opinion should only be performed in patients with severe, refractory hypoxemia [11]. Undoubtedly, these patients should be adequately monitored to ensure that intracranial pressure does not increase when the patient is placed in prone position. The administration of nitric oxide by inhalation should be considered with caution before being prescribed, because its use can increase the risk of hemorrhagic by compromising platelets function.
In patients with intracranial hypertension and NPE, the use of mannitol should be preferred to the administration of hypertonic saline solutions. To decrease the intracranial pressure, the induction of serum hyperosmolarity can accelerate the resolution of pulmonary edema [12]. Pharmacological neuromuscular paralysis can be useful to attain a better control of the intracranial pressure and may be necessary to optimize ventilation and oxygenation. Decompressive craniectomy is a valid option in refractory cases.
The hemodynamic therapy should be ideally guided by echocardiography and, sometimes, pulmonary catheterism. In most cases, bedside echocardiography obviates the need for pulmonary catheterism. When cardiac function is compromised, the use of inotropic drugs can be efficacious. Dobutamine, milrinone, and dopamine (in moderate doses) are valid options. Depending on the systemic arterial pressure, vasodilator drugs (e.g. nicardipine or clonidine), or less frequently vasoconstrictors (e.g. norepinephrine) may be necessary. It is recommended to avoid the use of beta-blockers in all the cases where there is evidence or suspicion of severe heart failure. However, in less severe cases I tend to favor the use of moderate doses of a beta-blocker to prevent additional adrenergically-induce myocardiac injury. Diuretics, such as furosemide, can be used to decrease the preload and they can be administered in combination with mannitol. Once hypoxemia has been corrected, it is very important to remember that in patients with aSAH it is neither necessary nor recommended to maintain the intravascular space contracted, due to the negative effects of hypovolemia on the risk of cerebral ischemia from vasospasm [2]. Once the NPE has resolved, there is no contraindication in these patients for the induction of hemodynamic augmentation therapy (induced hypertension and moderate hypervolemia) if they develop symptomatic vasospasm.
86.2.5 Prognosis
The occurrence of NPE has been associated with worsened functional prognosis, particularly in patients with aSAH [1-3]. This association may be explained by the greater severity of the primary neurological insult in those patients who develop MSE as a complication. Patients with NPE or other acute oxygenation problems often need more prolonged hospitalization in the intensive care unit [5]. However, NPE is eminently treatable and it usually resolves within 24-48 hours with adequate therapy [1].
86.3 Neurogenic Acute Cardiomiopathy
86.3.1 Features, Causes and Incidence
Different cardiac derangements can occur in association with cerebral diseases, including electrocardiographic changes, elevation of cardiac enzymes and transient left ventricular dysfunction. While the majority of these canges are not specific, there is a characteristic form of neurogenic cardiomyopathy known as tako-tsubo or apical ballooning syndrome [13]. This cardiac disorder is usually caused by acute physiological (e.g. in the case of intracranial hemorrhages) or emotional (e.g. “voodoo death”) stressors, hence the alternative denomination of “stress-induced cardiomyopathy” [14].
The most frequent electrocardiographic changes are seen during repolarization: T waves inversion (known as cerebral T waves), increase of the QTc interval, and appearance of U waves (Figure 86.3).
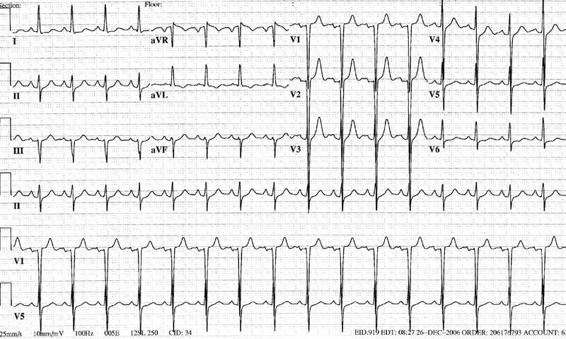
Figure 86.3. Example of electrocardiographic changes in a patient with subarachnoid hemorrhage.
ST segment elevation can be confused with acute myocardial ischemia. Tansient Q waves that do not follow the standard evolution of ischemic cases can occur, but are less frequent. These changes were initially described in patients with aSAH and these are the patients in whom they are most often observed in clinical practice. On the basis of the criteria used to define them, the incidence of EGC abnormalities reported in different studies varies, but it is usually accepted that they occur in 40-70% of aSAH cases [13]. The incidence is lower, but nonetheless elevated (15-40%), in patients with ischemic stroke [13].
Cardiac arrhythmias are frequent too. Benign arrhythmias are most common, but fatal arrhythmias can occur. The spectrum of possible arrhythmias includes atrial or ventricular extrasystole, supraventricular tachycardia, torsade de pointes and ventricular tachycardia or ventricular fibrillation [13,15]. In ischemic strokes, the compromise of the insula increases the risk of ventricular arrhythmias and sudden death [16,17]. Whether the side of insular involvement affects this risk is still under discussion [16,17].
Elevation of cardiac enzymes is frequent in patients with cerebrovascular events, especially if they are severe. Elevated cardiac enzymes can also be seen in patients with seizures, encephalitis, or brain trauma. The levels of both CK-MB and troponin can increase [13,18]. Characteristically they reach a peak during the first day and start declining over the following 1 to 4 days. They correlate with electrocardiographic and echocardiographic changes, but not with coronary disease [19]. Blood levels of brain natriuretic peptide (BNP) are usually increased in patients with electrocardiographic abnormalities [20].
Neurogenic cardiomyopathy can exhibit characteristic echocardiographic features. Regional abnormalities in ventricular motility do not respect the distribution of one coronary artery. Most often involve the apical and septal ventricular segments become akinetic or hypokinetic with relative preservation or hyperkinesis of the basal segments [21]. The resulting appearance of the heart on the ventriculogram explains why this condition has been named tako-tsubo cardiomyopathy (“Tako-tsubo” is the Japanese name for octopus traps that fishermen still use to catch octopus) or apical ballooning syndrome. Yet, atypical echocardiographic patterns can also occur [22]. This kind of cardiomyopathy is more frequent in post-menopausal women suffering of severe aSAH [14,19,23]. In patients with ischemic stroke, this pathology is much more frequent in women with stroke and insular damage [24]. Ejection fraction of the left ventricle decreases suddenly after the cerebral event, but returns to normal over the following 4 weeks [13,14,23]. It can be associated with acute pulmonary edema and can require hemodynamic support.
86.3.2 Pathophysiology
Acute neurogenic cardiomyopathy is thought to be caused by the injury of myocardial fibers and terminal nerves due to a massive sympathetic discharge [13,14,25,26]. This pathophysiological explanation is supported by the reports of apical ballooning in after infusion of cathecolamines [27]. In patients with SAH-related cardiac dysfunction a state of sympathetic denervation with normal myocardial perfusion has been demonstrated [25]. The pathological correlate is contraction band necrosis (also known as myofibrillary degeneration or coagulative myocytolysis) that occurs in proximity of sympathetic nerve terminals and does not respect the boundaries of coronary artery territories [13].
86.3.3 Diagnostic Evaluation
All patients with acute cerebral injuries should be managed in intensive care units, ideally dedicated units with neurological expertise, and be kept on cardiac telemetry. The initial evaluation should include an electrocardiogram, a chest X-ray and cardiac enzymes in all the cases, and a transthoracic electrocardiogram in patients with pulmonary edema, hypotension, or other manifestations of cardiac dysfunction. Furthermore it is prudent to perform an echocardiogram in postmenopausal women with severe aSAH even if there are no signs of cardiac failure, because these patients are at high risk of developing stress-induced cardiomyopathy. In patients with echocardiographic changes compatible with neurogenic cardiomyopathy, it is not necessary to evaluate the coronary circulation. In these cases it is advisable to repeat an echocardiogram after 1-2 weeks to reassess the ventricular function. Pulmonary catheterization is only necessary in cases of refractory shock. Figure 86.4 highlights a diagnostic algorithm to be considered when acute neurocardiogenic injury is suspected.
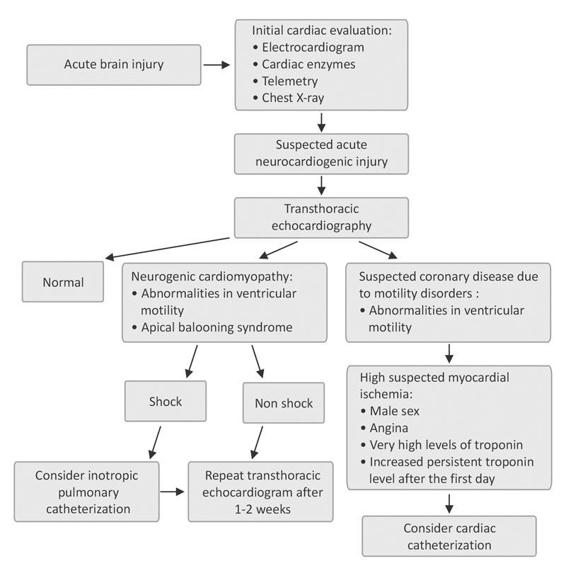
Figure 86.4. Evaluation and management of patients with suspected acute neurogenic cardiomyopathy. Modified from [14].
86.3.4 Treatment
The main tenets of treatment of acute neurogenic cardiomyopathy are hemodynamic support and cardiac rhythm monitoring. It is preferable to use inotropic drugs instead of pure vasopressors in case of hypotension. Diuretics such as furosemide are necessary when cardiogenic pulmonary edema is present. Arrhythmias should be treated as in other cases. In patients with prolonged QTc interval it is particularly important to avoid or correct any degree of hypokalemia and hypomagnesemia. In patients with aSAH and vasospasm who still have depressed cardiac function, therapeutic options include inotropic drugs, insertion of an intra-aortic balloon pump, and intra-arterial techniques, such as angioplasty or selective infusion of calcium channel blockers [13,14].
The prophylactic use of beta-blockers in patients at high risk of acute neurogenic cardiomyopathy can be useful, but more studies are needed before it can be recommended.
86.3.5 Prognosis
Although the cardiac dysfunction is reversible [28], most studies have shown that the presence of elevated troponin levels and electrocardiographic changes are associated with worse prognosis in patients with aSAH, intraparenchymal hemorrhage and ischemic stroke [18,29,30]. Available data is insufficient to determine if this correlation could be a direct consequence of cardiac damage or a surrogate marker of more severe acute brain disease.
References
1. Baumann A, Audibert G, McDonnell J, et al. Neurogenic pulmonary edema. Acta Anaesthesiol Scand 2007; 51: 447-55
2. Friedman JA, Pichelmann MA, Piepgras DG, et al. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery 2003; 52: 1025-31
3. Muroi C, Keller M, Pangalu A, et al. Neurogenic pulmonary edema in patients with subarachnoid hemorrhage. J Neurosurg Anesthesiol 2008; 20: 188-92
4. Solenski NJ, Haley EC Jr., Kassell NF, et al. Medical complications of aneurysmal subarachnoid hemorrhage: a report of the multicenter, cooperative aneurysm study. Participants of the Multicenter Cooperative Aneurysm Study. Crit Care Med 1995; 23: 1007-17
5. Vespa PM, Bleck TP. Neurogenic pulmonary edema and other mechanisms of impaired oxygenation after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2004; 1:157-70
6. Smith WS, Matthay MA. Evidence for a hydrostatic mechanism in human neurogenic pulmonary edema. Chest 1997; 111: 1326-33
7. Theodore J, Robin ED. Pathogenesis of neurogenic pulmonary edema. Lancet 1975; 2: 749-51
8. McClellan MD, Dauber IM, Weil JV. Elevated intracranial pressure increases pulmonary vascular permeability to protein. J Appl Physiol 1989; 67: 1185-91
9. Mayer SA, Fink ME, Homma S, et al. Cardiac injury associated with neurogenic pulmonary edema following subarachnoid hemorrhage. Neurology 1994; 44: 815-20
10. McGuire G, Crossley D, Richards J, et al. Effects of varying levels of positive end-expiratory pressure on intracranial pressure and cerebral perfusion pressure. Crit Care Med 1997; 25: 1059-62
11. Marshall SA, Nyquist P. A change of position for neurogenic pulmonary edema. Neurocrit Care 2008
12. Toung TJ, Chang Y, Lin J, et al. Increases in lung and brain water following experimental stroke: effect of mannitol and hypertonic saline. Crit Care Med 2005; 33: 203-8
13. Kopelnik A, Zaroff JG. Neurocardiogenic injury in neurovascular disorders. Crit Care Clin 2006; 22: 733-52
14. Lee VH, Oh JK, Mulvagh SL, et a. Mechanisms in neurogenic stress cardiomyopathy after aneurysmal subarachnoid hemorrhage. Neurocrit Care 2006; 5: 243-9
15. Cheung RT, Hachinski V. Cardiac effects of stroke. Curr Treat Options Cardiovasc Med 2004; 6: 199-207
16. Ay H, Koroshetz WJ, Benner T, et al. Neuroanatomic correlates of stroke-related myocardial injury. Neurology 2006; 66: 1325-9
17. Laowattana S, Zeger SL, Lima JA, et al. Left insular stroke is associated with adverse cardiac outcome. Neurology 2006; 66: 477-83
18. Jensen JK, Kristensen SR, Bak S, et al. Frequency and significance of troponin T elevation in acute ischemic stroke. Am J Cardiol 2007; 99: 108-12
19. Tung P, Kopelnik A, Banki N, et al. Predictors of neurocardiogenic injury after subarachnoid hemorrhage. Stroke 2004; 35: 548-51
20. Tung PP, Olmsted E, Kopelnik A, et al. Plasma B-type natriuretic peptide levels are associated with early cardiac dysfunction after subarachnoid hemorrhage. Stroke 2005; 36: 1567-19
21. Tsuchihashi K, Ueshima K, Uchida T, et al. Transient left ventricular apical ballooning without coronary artery stenosis: a novel heart syndrome mimicking acute myocardial infarction. Angina Pectoris-Myocardial Infarction Investigations in Japan. J Am Coll Cardiol 2001; 38: 11-8
22. Hurst RT, Prasad A, Askew JW 3rd, et al. Takotsubo. cardiomyopathy: a unique cardiomyopathy with variable ventricular morphology. JACC Cardiovasc Imaging 2010; 3: 641-9
23. Lee VH, Connolly HM, Fulgham JR, et al: Tako-tsubo cardiomyopathy in aneurysmal subarachnoid hemorrhage: an underappreciated ventricular dysfunction. J Neurosurg 2006; 105: 264-70
24. Yoshimura S, Toyoda K, Ohara T, et al. Takotsubo cardiomyopathy in acute ischemic stroke. Ann.Neurol 2008; 64: 547-54
25. Banki NM, Kopelnik A, Dae MW, et al. Acute neurocardiogenic injury after subarachnoid hemorrhage. Circulation 2005; 112: 3314-9
26. Lindsay J, Paixao A, Chao T, et al. Pathogenesis of the Takotsubo syndrome: a unifying hypothesis. Am J Cardiol 2010; 106: 1360-3
27. Abraham J, Mudd JO, Kapur N, et al. Stress cardiomyopathy after intravenous administration of caecholamines and beta receptor agonists. J Am Coll Cardiol 2009; 53: 1320-5
28. Madhavan M, Rihal CS, Lerman A, et al. Acute heart failure in apical ballooning syndrome (TakoTsubo/stress cardiomyopathy): clinical correlates and Mayo Clinic risk score. J Am Coll Cardiol 2011; 57: 1400-1
29. Hays A, Diringer MN. Elevated troponin levels are associated with higher mortality following intracerebral hemorrhage. Neurology 2006; 66: 1330-4
30. van dB, I, Hasan D, Vandertop WP, et al. Impact of cardiac complications on outcome after aneurysmal subarachnoid hemorrhage: a meta-analysis. Neurology 2009; 72: 635-42
Related posts:
 Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
Neuroscience Critical Care: Two Experts’ Point of View
Neurocritical Care of Patients With Arteriovenous Malformations
 Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
Pathophysiology of Intracranial Hypertension
Concepts and Management of Brain Death and Management of Potential Organ Donation
 Monitoring Brain Chemistry by Microdialysis During Neurointensive Care
Monitoring Brain Chemistry by Microdialysis During Neurointensive Care
 Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Acute Paraplegias and Quadriplegias of Non-traumatic Cause
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


