Neuropathology of Developmental Disorders Associated with Epilepsy
Harry V. Vinters
Noriko Salamon
Hajime Miyata
Negar Khanlou
Gary W. Mathern
Introduction
Structural lesions associated with epilepsy can be categorized into the following groups: (a) malformative, (b) neoplastic, (c) familial/metabolic, (d) vascular/traumatic, (e) infectious/inflammatory, and (f) associated with Ammon’s horn (hippocampal, mesial temporal) sclerosis.44,184 Several authors have suggested that some of these pathologic processes may overlap in epilepsy patients,12,16 and dual pathology (e.g., a corticectomy showing features of both a malformative and destructive lesion) may occasionally be encountered.116 Some of the causal lesions are amenable to definitive surgical treatment (e.g., low-grade neoplasms), whereas others (e.g., metabolic diseases) frequently undergo diagnostic brain biopsy, sometimes unintentionally. This chapter focuses on developmental malformations of the neocortex that account for the majority of structural lesions seen in infants and children with intractable epilepsy, especially those with infantile spasms (IS). (The other categories of epileptogenic structural lesions are discussed elsewhere.) More subtle malformations are also recognized with increasing frequency in adults with epilepsy.
Neocortical malformations have usually been considered to represent developmental disorders of neuronal migration (or vascular formation), and have variously been classified with regard to morphology or putative etiology.8,135,138 The categorization of these heterogeneous lesions is increasingly being modified by (a) the availability of high-resolution multimodality neuroimaging techniques that can be used to predict pathologic abnormalities in a given patient24,26,158 and (b) molecular genetic clues to pathogenesis based on techniques such as gene expression profiling following laser capture microdissection of surgical specimens.32 The genetic basis of some types of lissencephaly (e.g., Miller-Dieker syndrome) is now quite well understood.32,147 They can be roughly categorized as follows: (a) cortical dysplasia (CD), which accounts for the majority of malformations associated with pediatric epilepsy and encompasses the full spectrum of neuronal migration disorders (NMDs), sometimes also described as malformations of cortical development (MCDs), ranging from the most subtle to the most severe; (b) structural lesions associated with tuberous sclerosis complex (TSC); (c) Sturge-Weber-Dimitri syndrome (SWDS), also known as encephalotrigeminal angiomatosis; (d) neurofibromatosis type 2, which may be associated with meningio-angiomatosis; and (e) vascular malformations.
This chapter discusses clinicopathologic associations between the recognized pediatric epilepsy syndromes and their neuroradiologic and neuropathologic substrates. We review the terminology used to characterize these complex malformative lesions in the light of modern neuroimaging data. We give a brief overview of development of the cerebral cortex and identify the points of susceptibility at which the error or errors (genetic or environmental) resulting in cortical malformations may occur.
Clinicopathologic Features
Epilepsy has traditionally been classified into syndromes based on clinical presentation and electroencephalographic (EEG) findings. However, there is often a marked discrepancy between the clinical phenomenology of a seizure disorder and its neuropathologic substrate(s). Infantile spasms (IS, West syndrome) and Lennox-Gastaut syndrome can be seen with a wide range of cerebral lesions,69,106,153 indicating the nonspecific nature of these seizure disorders. The clinical form of epilepsy seen in a given patient appears to depend more on when during cerebral development the lesion occurred than on the specific type or topographic distribution of lesions.69,108 It has been suggested that the central nervous system (CNS) lesions associated with IS can be functionally categorized into three groups: (a) diffuse, (b) focal or multifocal cerebral lesions, and (c) cases with minimal neuropathologic change.153 Diffuse hemispheric lesions include hemimegalencephaly (HME), agyria/pachygyria-lissencephaly, and Aicardi syndrome.1 Focal and multifocal lesions include CD and destructive lesions (vascular and infectious), as well as cortical tubers seen in patients with TSC.
Developmental malformations of the neocortex (malformations of cortical development, MCDs) can be considered a spectrum of CD resulting from derangement of the normal process of cortical development.117,120,130,131,142 This spectrum consists of a range of morphologic features associated with multiple putative etiologic factors, including genetic and environmental (e.g., destructive) influences. CD encompasses the full spectrum of neocortical malformations, ranging from the most subtle (microdysgenesis) to the most severe (HME), and includes such conditions as agyria/pachygyria-lissencephaly, polymicrogyria (PMG), and focal CD. CD, therefore, comprises a spectrum of derangements in neocortical development that are associated with a range of morphologic features and with multiple putative etiologic factors, including genetic and environmental influences. The resultant neuropathologic features may reflect abnormalities that probably occur within discrete time windows during brain development. Clinically there is an inverse correlation between the size and severity of CD and the age at clinical presentation,26,117 supporting the notion that there is a predominance of pathologically severe CD in neonates and infants with seizures, including those
with IS.73 Although CD accounts for the majority of malformations associated with pediatric epilepsy, other malformative lesions are also capable of producing these epilepsy syndromes, and they frequently show neuropathologic changes that overlap significantly with those seen in patients with CD.
with IS.73 Although CD accounts for the majority of malformations associated with pediatric epilepsy, other malformative lesions are also capable of producing these epilepsy syndromes, and they frequently show neuropathologic changes that overlap significantly with those seen in patients with CD.
Jellinger69 noted that the clinical severity of a given seizure disorder appears to be more closely related to the timing of the insult (whether genetic or environmental) and its effect on the processes of development than to the nature of the lesion itself. This may explain some of the heterogeneity of neuropathologic lesions seen in children with epilepsy because the clinical phenotype results not only from the lesion or putative insult to the developing CNS, but also from the developmental processes it affects.
A puzzling feature noted in pediatric epilepsy is the finding in cases of IS of diffuse and transient suppression of cortical activity in the presence of focal or multifocal lesions.153 This diffuse cortical suppression has not been linked to particular distributions or types of neuropathologic change. Rather, Robain and Vinters suggested that this feature of IS is associated with neocortical immaturity.153 The pattern of evolution of IS from transient and diffuse suppression of cortical discharge to ultimately focal or generalized seizures may represent a maturation of the malformed cortex.153
Nosology
The nosology and understanding of the full spectrum of CD are evolving and reflect a progressive elucidation of the basis of these extremely complex lesions. Initially, cortical malformations were referred to largely by their gross characteristics (i.e., agyria/pachygyria-lissencephaly, HME, microgyria). As investigators discovered the range of microscopic cortical malformations that produce epilepsy but show no (or relatively mild) gross abnormalities, additional terms such as microdysgenesis,57,107,108 dysplastic cortical architecture (not otherwise specified),157 focal cortical dysplasia,119,174 and generalized or diffuse cortical dysplasia78,83,96 were added to the literature. The nomenclature of CD has evolved through several schema of classification, each intended to provide correlations with morphology of seizures and reflect etiologic mechanisms.8,9,117 Focal cortical dysplasia was initially used to specify lesions in which cytomegalic neurons were present,174 although its frequent use to refer to a localized region of CD renders the term somewhat ambiguous. Some investigators prefer to use a traditional classification of migrational disorders into four main groups: (a) agyria/pachygyria-lissencephaly, (b) microgyria-polymicrogyria, (c) dysplastic cortical architecture, and (d) heterotopias.157 Others, to denote the belief that these lesions are related and reflect developmental abnormalities along a continuous spectrum, have chosen to refer to this group of lesions as neuronal migration disorders or NMDs,8,130,131 cerebral dysgenesis,159 or synaptic dysgenesis.13 The nosology of developmental neocortical malformations will continue to evolve, especially in the era of high-resolution magnetic resonance imaging (MRI), and reflect a progressive understanding of the underlying biology of these complex lesions (see Chapter 259). A recent consensus conference resulted in a proposal to subclassify CD or grade its severity (in surgical resection specimens) using simple morphologic criteria identifiable by any experienced neuropathologist, for example, presence or absence of cortical disorganization, enlarged or dysmorphic neuronal cell bodies, and “balloon cells” (see later discussion).132 This classification scheme has been adopted widely enough that surgical pathology reports on corticectomy specimens now frequently contain the terminology “Cortical dysplasia, Palmini type….”
Development of the Neocortex
Overview of Neocortical Development
The neuropathologic changes seen in children with epilepsy frequently represent the end results of insults to a rapidly developing brain. This section briefly summarizes the normal process of neocortical development and identifies the points of susceptibility at which the error or errors (genetic, environmental, or both) result in cases of pediatric epilepsy—usually due to CD. We also briefly introduce some recent advances in the genetic and molecular mechanisms that regulate cortical development. Many excellent reviews have summarized the historical evolution in our understanding of the complex neurobiologic, cellular, and molecular processes that work in concert to create a normally functioning cerebral cortex.30,52,67,141,160 Neocortical development after neural tube formation can roughly be considered to be the result of a series of overlapping processes: (a) cell proliferation in the ventricular zone and subventricular zone (VZ/SVZ), (b) early differentiation of neuroblasts and glioblasts, (c) programmed cell death of neuronal precursors and neurons, (d) migration of neuroblasts to form the cortical plate, (e) late neuronal migration, (f) organization and maturation of the cortex, and (g) synaptogenesis.1,8,157,175 Abnormalities of these processes result in abnormalities of cortical architecture and, by inference, its electrophysiologic properties. Most developmental disorders of the brain commonly associated with epilepsy are believed to originate from the perturbation of developmental events after the embryonic period, i.e., after 6 weeks’ gestation, when cell proliferation starts along the wall of the neural tube. This generates a collection of matrix cells,45 or precursor cells for all neuroblasts and glioblasts, forming ventricular and subventricular zones (VZ/SVZ) in the pallium, as well as the ganglionic eminence in the subpallium (Table 1).
Table 1 Major stages of human central nervous system development and associated epileptic disorders | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Programmed cell death (PCD) is an essential mechanism for normal brain development, determining the size and shape of the nervous system. In normal brain development, there is a 25% to 50% overproduction of neuroblasts; these undergo physiologic PCD.159 Neuroblasts and glia undergo this process as well. PCD appears to be under tight genetic control, as demonstrated in the Caenorhabditis elegans model.39 It is an active process that can be blocked by inhibitors of protein synthesis and RNA transcription.70 Failure of PCD may lead to mechanical barriers to migration and abnormal numbers of neurons. Supplemental to PCD, there is a conspicuous elimination of synapses occurring during development that is essential to remodeling of the cortex.140 This may be accomplished by different mechanisms, and competition for trophic substances has been suggested as one. Synapse elimination is highly intertwined with the remodeling of cortical connections and is a highly dynamic process140 demonstrated both in vivo and in vitro.180
Terminal differentiation of a neuroblast appears to be a multistep process.22,102,128 Cell surface properties and extracellular matrix molecules play a crucial role in influencing migration and terminal differentiation of neuroblasts. Neural adhesion and migration are governed by a series of morphoregulatory molecules, cell-adhesion molecules (CAMs), and substrate-adhesion molecules (SAMs). CAMs
are transmembrane molecules bearing extracellular domains that are homologous to domains of the immunoglobulin superfamily38 and are engaged in homotypic binding. The binding functions interact with the cytoskeleton because CAMs are transmembrane molecules. These molecules are involved in cell adhesion, axon binding, growth cone interactions, and other migrational mechanisms. SAMs, extracellular matrix molecules, are involved in the regulation of cell shape, motion, and process extension.36 These molecules are expressed in a spatial-temporal fashion and are under the control of homeobox-containing genes that are known to govern place-dependent morphology.74,75 The time-dependent levels of expression of the molecules are characteristic of a given anatomic area but are dynamically regulated and subject to local influences. The activities of these molecules are further modulated by neural activity itself.37 These morphoregulatory molecules can mediate neuron–neuron interactions (NCAMs), neuron–glia interactions (Ng-CAMs), or neuron–extracellular matrix interactions.30,60,61 Extracellular matrix molecules also appear to be important in cell motion, attraction, repulsion, and growth cone migration; soluble trophic factors have an important role in these processes.34
are transmembrane molecules bearing extracellular domains that are homologous to domains of the immunoglobulin superfamily38 and are engaged in homotypic binding. The binding functions interact with the cytoskeleton because CAMs are transmembrane molecules. These molecules are involved in cell adhesion, axon binding, growth cone interactions, and other migrational mechanisms. SAMs, extracellular matrix molecules, are involved in the regulation of cell shape, motion, and process extension.36 These molecules are expressed in a spatial-temporal fashion and are under the control of homeobox-containing genes that are known to govern place-dependent morphology.74,75 The time-dependent levels of expression of the molecules are characteristic of a given anatomic area but are dynamically regulated and subject to local influences. The activities of these molecules are further modulated by neural activity itself.37 These morphoregulatory molecules can mediate neuron–neuron interactions (NCAMs), neuron–glia interactions (Ng-CAMs), or neuron–extracellular matrix interactions.30,60,61 Extracellular matrix molecules also appear to be important in cell motion, attraction, repulsion, and growth cone migration; soluble trophic factors have an important role in these processes.34
Radial Migration of Neuroblasts from the Subventricular Zone
Radial migration refers to the process by which neuroblasts from the VZ/SVZ migrate along the processes of radial glia to reach the (neo)cortex. Radial glia initially have cell bodies in the ventricular zone and end-feet on the pial surface; with time, they detach themselves from the ventricular lining and migrate toward the cortex.32a,41 They function as a permissive scaffold on which neuroblasts may migrate from the ventricular zone to the cortical plate.60 At approximately 4 weeks’ gestation, the neural tube forms with a simple pseudostratified neuroepithelium, component cells of which then proliferate around the developing ventricular system, eventually becoming the ventricular zone. The ventricular zone can be described as a mosaic of precursors that will give rise to neurons, astrocytes, and oligodendroglia, as demonstrated using retroviral markers.139 The first postmitotic cells form the preplate or primordial plexiform layer98,110,112,168,194 above the ventricular zone and beneath the pia. Toward the end of the embryonic period, cells from the ventricular zone migrate to form the cortical plate within the preplate; that is, the cortical plate is formed dividing
the preplate (preplate splitting) into (a) a thin superficial component, the marginal zone, and (b) a thick, deeper component, the subplate. The intermediate zone or future subcortical white matter also appears by this stage,157 and this cortical plate formation continues until 16 weeks’ gestation.9 The neocortex is formed in an “inside-out” fashion: Neuroblasts born first, destined for the deepest cortical layers, migrate first, whereas neuroblasts born later, destined for the more superficial cortical layers, migrate past the already present cortical neuroblasts,6 eventually forming a six-layered structure.163 Even after this point, however, neuroblasts continue to migrate through the intermediate zone (future white matter) to the cortex, a process that can continue up to a few months after birth.163 Abnormalities of radial glia may occur through various molecular mechanisms either focally or diffusely in malformations of cortical development (MCDs), including Fukuyama congenital muscular dystrophy (FCMD)169 and tuberous sclerosis complex (TSC).133 Alterations of signaling pathways that regulate microtubule dynamics either directly or indirectly may also result in a derangement of radial glial fibers, as has been demonstrated in reeler mice64 and Lis1 mutant mice.19 Radial glia have long been known to serve as guides for migrating neuroblasts and finally give rise to cortical astrocytes. However, recent evidence in mice indicates that radial glial cells also generate neurons in the developing cerebral cortex. Three distinct subsets are thus identified, including subpopulations important in gliogenesis, neurogenesis, and both.7,58
the preplate (preplate splitting) into (a) a thin superficial component, the marginal zone, and (b) a thick, deeper component, the subplate. The intermediate zone or future subcortical white matter also appears by this stage,157 and this cortical plate formation continues until 16 weeks’ gestation.9 The neocortex is formed in an “inside-out” fashion: Neuroblasts born first, destined for the deepest cortical layers, migrate first, whereas neuroblasts born later, destined for the more superficial cortical layers, migrate past the already present cortical neuroblasts,6 eventually forming a six-layered structure.163 Even after this point, however, neuroblasts continue to migrate through the intermediate zone (future white matter) to the cortex, a process that can continue up to a few months after birth.163 Abnormalities of radial glia may occur through various molecular mechanisms either focally or diffusely in malformations of cortical development (MCDs), including Fukuyama congenital muscular dystrophy (FCMD)169 and tuberous sclerosis complex (TSC).133 Alterations of signaling pathways that regulate microtubule dynamics either directly or indirectly may also result in a derangement of radial glial fibers, as has been demonstrated in reeler mice64 and Lis1 mutant mice.19 Radial glia have long been known to serve as guides for migrating neuroblasts and finally give rise to cortical astrocytes. However, recent evidence in mice indicates that radial glial cells also generate neurons in the developing cerebral cortex. Three distinct subsets are thus identified, including subpopulations important in gliogenesis, neurogenesis, and both.7,58
Tangential Migration of GABAergic Interneurons From Ganglionic Eminence
Tangential migration refers to the process by which neuroblasts from the ganglionic eminence migrate in a nonradial, “neurophilic” fashion, possibly along neuronal processes rather than radial glial fibers, moving tangential to the pial surface to form complex three-dimensional neural structures.143 Ganglionic eminence, primarily considered to be a source of basal ganglia neurons, consists of three parts. The medial ganglionic eminence, derived from the diencephalon, gives rise to globus pallidus. The lateral ganglionic eminence, derived from the telencephalon, gives rise to the caudate nucleus and putamen. The caudal ganglionic eminence primarily gives rise to amygdala. Both lateral and median ganglionic eminences, however, are also involved in the formation of cerebral cortex. In fact, mechanically separating the ganglionic eminence from the cortex results in a loss of calbindin and γ-aminobutyric acid (GABA)–positive neurons in the cortex,2 and 35% of cortical GABAergic interneurons arise form both ganglionic eminences by way of tangential migration.3,87 This contrasts with the origin of pyramidal neurons of the cerebral cortex, which arise from the ventricular zone of the pallium by way of radial migration. Although 65% of cortical GABAergic interneurons arise from the ventricular zone of the pallium, they are considered to migrate in nonradial, neurophilic fashion at least within the VZ/SVZ.87 Migration of GABAergic interneurons seems to be much more complex, because “ventricle-directed migration” has also been demonstrated in mice.122 GABAergic interneurons are immunoreactive for calcium-binding proteins such as calbindin. Studies of macaque monkey brain have suggested that calcium-binding-protein–containing interneurons make up 90% of all GABAergic neurons in the cerebral cortex.91 The number and distribution of calcium-binding-protein–containing neurons in the neocortex can be reorganized in early postnatal life149; by 28 weeks after birth, the laminar distribution of calbindin-immunoreactive cells shifts from layer IV to layer II. It has been suggested that doublecortin (DCX) is important for nonradial migration because it is rather selectively expressed in tangentially oriented postmitotic neurons in the subventricular and intermediate zones.113
Origin of Cajal-Retzius Cells and Their Biologic Significance
The marginal zone, future molecular layer or cortical layer I, is composed largely of Cajal-Retzius cells. They secrete the extracellular glycoprotein reelin that is required for the normal inside-out positioning of neurons as they migrate from the ventricular zone along radial glia. Human Cajal-Retzius cells, characterized by the combined expression of reelin and p73, are transient cells, are present from the preplate stage at 8 weeks’ gestation, and gradually increase in number (by tangential migration) until they disappear by the end of gestation.110,113 One possible origin of Cajal-Retzius cells is considered to be the boundary between prospective hippocampus and choroid plexus epithelium or “cortical hem” in the dorsal telencephalon.113 In mice carrying mutations in RELN (reeler mice) and in disabled-1 (Dab1) as well as in mice carrying double mutations of both very low density lipoprotein receptor (VLDLR) and apolipoprotein E receptor 2 (ApoER2), normal neuroblast migration with an inside-out fashion is inverted.178 This suggests a role for these genes in the control of cell positioning in the developing central nervous system and even predicts a pattern of cytoarchitectural alteration in patients carrying alterations in the reelin/lipoprotein receptor/Dab1 pathway, as well as RELN mutations causing lissencephaly.63 LIS1 also appears to have important effects on neuronal migration, and significant interactions with Cajal-Retzius cells: LIS1 deficiency negatively affects the migration and differentiation of both doublecortin- and reelin-positive neurons in the developing human brain.114
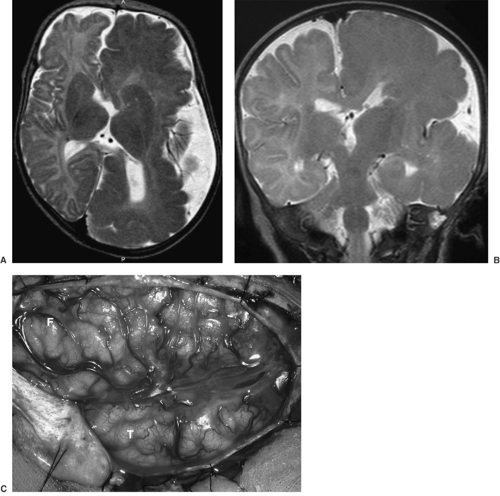 FIGURE 1. Hemimegalencephaly (HME) in a 6-month-old child. Axial (A) and coronal (B) T2-weighted magnetic resonance imaging (MRI) images show markedly enlarged left cerebral hemisphere with thickened gyri and smooth surface, consistent with HME. The left lateral ventricle and caudate head are deformed. White matter is diffusely hypointense compared with the unaffected right cerebral hemisphere, suggesting accentuated myelination. C: Intraoperative photograph of the region of HME oriented with the frontal lobe (F) in the top left and the temporal lobe (T) in the lower middle portions of the image. Notice the diffuse cortical disorganization of all gyri. This child had a monozygotic twin who was normal. (Panel B is from Salamon N, Andres M, Chute DJ, et al. Contralateral hemimicrencephaly and clinical-pathological correlations in children with hemimegalencephaly. Brain. 2006;129:352–365; with permission of the editor of Brain and Oxford University Press.) |
Origin of Superficial Granular Layer and Its Biologic Significance
The superficial or subpial granular layer (SGL) is a transient cell layer and appears beneath the pial surface between 13 and 24 weeks’ gestation.18,44 Cells in the SGL originate from the basal periolfactory subventricular zone18,46,111 and migrate tangentially beneath the pia to cover the neocortical marginal zone. Cells in this layer express interneuron markers such as calretinin, calbindin, and GABA,145 suggesting that they are equivalent to GABAergic interneurons. The biologic significance of the SGL, however, remains to be elucidated. Programmed cell death may, at least in part, contribute to elimination of the SGL.166 There is controversy in this research area because the SGL is also suggested to be an additional source of cortical interneurons; it disappears probably as the result of inward or ventricle-directed migration of its component cells.122,144,145
Subplate Neurons as Pioneer Cells to Form Early Thalamocortical Projections
The human subplate contains large multipolar neurons. Subplate neurons in the developing cerebrum, although they are transient and most disappear in early postnatal life, are believed to be important in organizing cortical connections in the developing cerebrum. They are believed to act as pioneer corticofugal axons.29,49,50,101
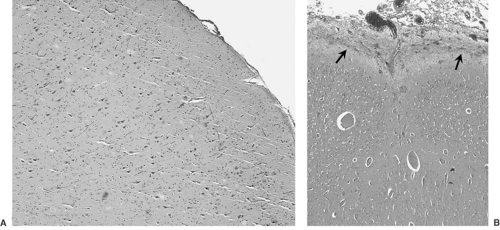 FIGURE 2. Representative micrographs of the resection specimen obtained from the child with magnetic resonance imaging (MRI) and intraoperative appearances illustrated in Fig. 1. A: Low-magnification view of a representative region of cortex shows a modest degree of neuronal disorganization. B: A focus of polymicrogyria, one of many seen in the resection specimen. Also note a “rind” of disorganized glial tissue covering the pia, with apparent extension into the subarachnoid space (arrows). (Hematoxylin and eosin stain.) (See the color insert.) |
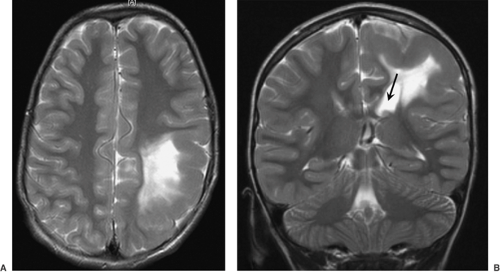 FIGURE 3. Focal cortical dysplasia extensively involving the left parietal lobe. T2-weighted axial (A) and coronal (B) images show thickened gyri and hyperintense signal in the white matter of the left parietal lobe. The left lateral ventricle is enlarged (arrow). |
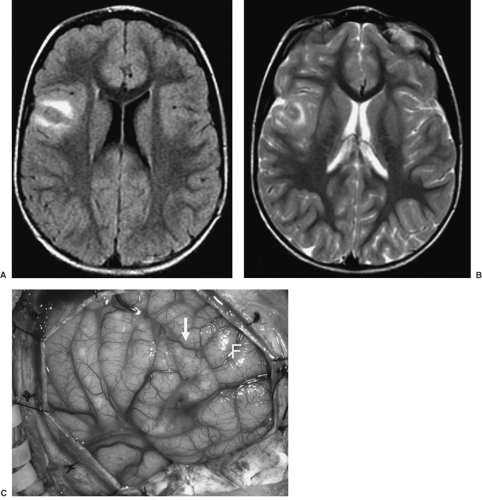 FIGURE 4. Axial FLAIR (fluid attenuated inversion recovery) (A) and T2-weighted axial (B) images demonstrate hyperintense signal area in the subcortical white matter of the right frontal operculum in a 4-year-old girl. The gray matter in this region is thickened, but there is no mass effect. The findings are suggestive of cortical dysplasia. C: View of the lesion intraoperatively (arrow). Histopathology of this lesion showed classic features of Palmini grade IIB cortical dysplasia. F, frontal lobe. |
Neuropathology and Pathophysiologic Significance of Cortical Dysplasia
The Pediatric Epilepsy Surgery program at UCLA Center For the Health Sciences, active since 1986, has enabled us to examine over 500 surgically resected specimens from infants and children with intractable seizures, ranging from partial lobectomies to complete and partial (functional) hemispherectomies. The most common morphologic substrate for this was CD, this being of etiologic importance as a cause of intractable pediatric seizures in >80% of children <3 years of age. The extent of CD neuropathology can be predicted by high-resolution neuroimaging studies. Such neuroimaging allows for stratification of CD cases into those that show hemimegalencephaly (HME), with diffuse enlargement of the gray and white matter, including thickening of the cortical ribbon, within an entire cerebral hemisphere (Figs. 1 and 2); hemispheric CD, with multifocal CD affecting one cerebral hemisphere (although not causing enlargement of that hemisphere); and multilobar, lobar, or focal CD (Figs. 3 and 4), the latter affecting as few as one or two adjacent gyri. HME is easily recognized by neuroimaging. The MRI findings include an enlarged cerebral hemisphere and markedly thickened gyri, with loss of sulcation. There is deformity and enlargement of the ipsilateral ventricle. Palmini type IIB CD is also easily identified by MRI, with focal thickness of the gyrus (gyri) and associated hyperintense T2-weighted signal changes in the adjacent white matter. It is difficult to visualize Palmini type I CD by MRI; however, on combining this with other modalities, such as positron emission tomography (PET) and magnetic source imaging (MSI), the detectability of this
lesion increases and foci of subtle gray–white matter blurring may be visualized intraoperatively.
lesion increases and foci of subtle gray–white matter blurring may be visualized intraoperatively.
Macroscopic heterotopia and polymicrogyria (PMG) are occasionally seen in resection specimens from such patients. Loss of the normal cortex–white matter junction, best appreciated with a Klüver-Barrera or other myelin stain, is a frequent accompaniment and excellent predictor of severe microscopic CD. Many specimens, however, exhibit no striking gross cortical abnormalities. CD can be further characterized with regard to specific and easily identifiable microscopic abnormalities,
which include cortical laminar disorganization (Fig. 5), single heterotopic white matter neurons, excess neurons in the neocortical molecular layer, marginal glioneuronal heterotopia, white matter neuronal heterotopia, neuronal cytomegaly with or without associated dysmorphic features of the cytoplasm (the latter almost invariably accompanied by cytoskeletal abnormalities) (Fig. 6), and balloon cell change (Fig. 7).117 These microscopic features can be used as the basis for a grading system for CD; one widely used schema is that presented recently by Palmini et al.132
which include cortical laminar disorganization (Fig. 5), single heterotopic white matter neurons, excess neurons in the neocortical molecular layer, marginal glioneuronal heterotopia, white matter neuronal heterotopia, neuronal cytomegaly with or without associated dysmorphic features of the cytoplasm (the latter almost invariably accompanied by cytoskeletal abnormalities) (Fig. 6), and balloon cell change (Fig. 7).117 These microscopic features can be used as the basis for a grading system for CD; one widely used schema is that presented recently by Palmini et al.132
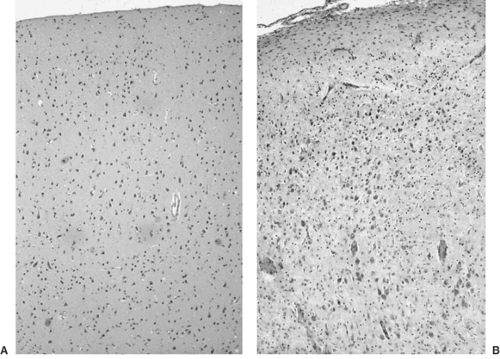 FIGURE 5. A: Relatively normally organized cerebral cortex from a surgical resection specimen. B: Region with severe cortical dysplasia shows pronounced cortical disorganization, with abnormal crowding of neurons and abnormal orientation of many cells. Both panels are from micrographs photographed at the same magnification from sections stained with routine hematoxylin and eosin stains. (See the color insert.) |
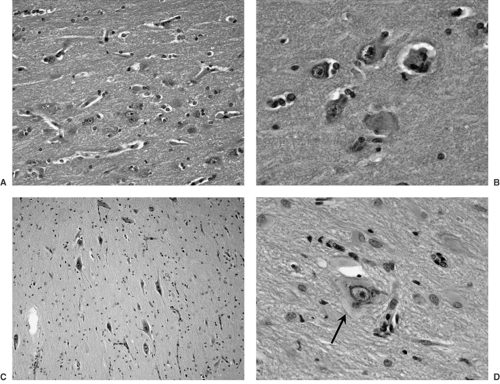 FIGURE 6. Neuronal disorganization and dysmorphism in cortical dysplasia/malformations of cortical development. A: Intermediate-magnification micrograph shows crowded and abnormally oriented neuronal cell bodies. B: Same features shown at a higher magnification. C: Variably enlarged and abnormally distributed neurons near the cortex–white matter junction. D: Magnified view of a dysmorphic neuron (arrow) with clumping of Nissl substance around the nucleus and clearing of the peripheral cytoplasm. Gemistocytic astrocytes are seen distributed around this neuron. (All panels are from hematoxylin and eosin–stained sections.) (See the color insert.) |
Cortical laminar disorganization is the most ubiquitous microscopic finding, obviously because it represents a defining histopathologic feature of CD. Because neocortical architecture is the end result of the developmental processes of proliferation of neuronal precursors, migration, terminal differentiation, PCD, and cortical remodeling (see earlier discussion), abnormalities in any of these processes may result in abnormal cortical architecture. Cortical disorganization (as well as cytologic abnormalities in individual neurons, especially when fairly subtle) can be highlighted using immunohistochemistry that incorporates primary antibodies to neurofilament epitopes (Fig. 8). Although many neurons still reside in the intermediate zone/white matter in the last trimester of pregnancy and even into postnatal life,80,163 the phenomenon of single heterotopic neurons in the white matter is accentuated in CD.108 It is present in the majority of our patients with CD, and has been demonstrated in other series using morphometric techniques.105 It has been suggested that injury to the radial glial fibers leads to a stranding of the migrating neuroblasts within the white matter, where they further differentiate into
mature neurons.159 Alternatively, overproduction of neurons late in neurogenesis may lead to crowding of migrating neurons toward the cortical surface.5 Morphometric analysis has also demonstrated a statistically significant increase in the number of neurons within the molecular layer of the cortex in epileptic patients versus controls,107 and this is considered to be evidence of a slight maldevelopment of the neocortex, sometimes described as microdysgenesis, although this term is now felt to be imprecise and best avoided.132 Persistence of the superficial granular layer (SGL) has been seen in association with many cortical malformations.117 Marginal glioneuronal heterotopia consists of excrescences of disorganized neuroglial tissue extending from the pial surface into the subarachnoid space. They are often found in association with persistent SGL and tend to occur in the same brain region as the other malformations. These lesions may be associated with a failure of the glia limitans.117 White matter neuronal heterotopia consists of disorganized masses of neurons in the white matter that usually occur in a periventricular position with a nodular morphology, although rare instances of laminar subcortical bands of heterotopic gray matter have been known to produce the appearance of a double cortex. It has been suggested that these are associated with injury to a group of radial glia, leading to failure of a group of neuroblasts to migrate.159 Alternatively, a defect in genes controlling neuroglial interactions, neuroblast proliferation, and PCD has been suggested as being causal.157
mature neurons.159 Alternatively, overproduction of neurons late in neurogenesis may lead to crowding of migrating neurons toward the cortical surface.5 Morphometric analysis has also demonstrated a statistically significant increase in the number of neurons within the molecular layer of the cortex in epileptic patients versus controls,107 and this is considered to be evidence of a slight maldevelopment of the neocortex, sometimes described as microdysgenesis, although this term is now felt to be imprecise and best avoided.132 Persistence of the superficial granular layer (SGL) has been seen in association with many cortical malformations.117 Marginal glioneuronal heterotopia consists of excrescences of disorganized neuroglial tissue extending from the pial surface into the subarachnoid space. They are often found in association with persistent SGL and tend to occur in the same brain region as the other malformations. These lesions may be associated with a failure of the glia limitans.117 White matter neuronal heterotopia consists of disorganized masses of neurons in the white matter that usually occur in a periventricular position with a nodular morphology, although rare instances of laminar subcortical bands of heterotopic gray matter have been known to produce the appearance of a double cortex. It has been suggested that these are associated with injury to a group of radial glia, leading to failure of a group of neuroblasts to migrate.159 Alternatively, a defect in genes controlling neuroglial interactions, neuroblast proliferation, and PCD has been suggested as being causal.157
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


