The most common parkinsonian disorders can be assigned to one of two categories based on biochemical and structural abnormalities in two major proteins in the central nervous system—tau protein or α-synuclein. (There is a family of synucleins [α-, β-, and γ-synuclein]; in this chapter, synuclein refers to α-synuclein.) The disease categories are termed “tauopathies” and “synucleinopathies” (1). Tau protein, the major structural component of neurofibrillary tangles (NFTs) in Alzheimer’s disease (AD), is a microtubule-associated protein that was initially considered exclusively neuronal, but tau-positive glial lesions are increasingly recognized in parkinsonian tauopathies (2). Mutations in the tau gene (MAPT), located on chromosome 17, and their relationship to frontotemporal dementia with parkinsonism (FTDP-17) (3), have led to renewed interest in tau and its role in parkinsonism, especially the parkinsonian tauopathies. Discovery of common genetic variants in MAPT in genome-wide association studies of idiopathic Parkinson’s disease (PD) (4), which is fundamentally a synucleinopathy, adds further evidence of a link between PD and tau. Synuclein was first found as a nonamyloid component of senile plaques in AD (5). Interest in synuclein was fueled by the discovery that its gene (SNCA), which is located on chromosome 4, was mutated in familial forms of PD (6,7). Shortly thereafter, synuclein was shown to be present in Lewy bodies (LBs) (8), as well as glial cytoplasmic inclusions (GCIs) in multiple system atrophy (MSA) (9).
Both tau and synuclein are heat-stable proteins with potential for protein–protein interactions (reviewed in (10)) with a tendency to self-associate to form pathologic fibrils; coding region mutations in each molecule favor fibril formation. The best characterized posttranslational modification of tau and synuclein is phosphorylation. Many protein kinases have been shown to phosphorylate tau, but only a limited set of kinases phosphorylate synuclein (reviewed in (11)). Phosphorylation of tau modifies its binding to tubulin, but little is known about the effects of phosphorylation of synuclein, except that this modification is an excellent marker for pathologic synuclein (12). Tau and synuclein in pathologic lesions are ubiquitinated (13,14). Although ubiquitination may merely be an adaptive cellular response, it may also have a role in the normal life cycle of these molecules and contribute directly to pathogenesis of PD.
Synuclein and tau inclusions in neurons and glia are neuropathologic hallmarks of major parkinsonian disorders. Abnormal tau accumulates in a range of glial inclusions, including oligodendroglial coiled bodies, tufted astrocytes, and astrocytic plaques (15). Synuclein is present in LBs and Lewy-like neuronal inclusions in MSA, as well as in GCIs in MSA and to a lesser extent in PD. Synuclein-positive oligodendroglial cytoplasmic inclusions are the sine qua non of MSA (16).
SYNUCLEINOPATHIES
LB DISORDERS: PD AND DEMENTIA WITH LEWY BODIES
In this discussion, PD and dementia with Lewy bodies (DLB) are used to refer to clinical syndromes with characteristic signs and symptoms, whereas Lewy body disease (LBD) is used as overarching term for the disease process associated with LBs. The cardinal clinical features of PD are bradykinesia, rigidity, tremor, and postural instability. The most common pathologic finding in PD is LBD, but occasionally progressive supranuclear palsy (PSP), MSA, or other disorders can mimic PD. The core clinical features of DLB are dementia, extrapyramidal signs, visual hallucinations, and a fluctuating course (17). The pathologic findings in DLB include LBs and varying degrees of Alzheimer-type pathology. Overlap between these two clinical syndromes is vexing in that dementia can occur in the course of PD (18) and some patients with DLB have little or no parkinsonism (19). A Movement Disorder Society task force has addressed the main unresolved issues (20).
LB and Lewy Neurites
The classic LB is a concentric hyaline cytoplasmic inclusion (Fig. 25.1A) in specific vulnerable populations of neurons. In some regions of the brain, such as the dorsal motor nucleus of the vagus and the basal nucleus of Meynert, similar inclusions occur within neuronal processes as intraneuritic LBs (Fig. 25.1H). Intraneuritic LBs can be detected in routine histopathologic preparations and should be distinguished from Lewy neurites, which are not visible with routine histology. Lewy neurites were first described in the hippocampus with ubiquitin immunohistochemistry (21), but are also found in other regions of the brain with synuclein immunohistochemistry, most often in amygdala, cingulate gyrus, and temporal cortex (Fig. 25.1F).
Ultrastructural studies of LBs demonstrate non–membrane-bound, granulofilamentous structures. The central region of the classical LB is usually amorphous dense material, whereas the periphery has radially arranged 10-nm-diameter filaments (22). At the electron microscopic level, Lewy neurites also have 10-nm filaments, but they lack a dense core, and the filaments are arranged haphazardly (21).
In contrast to classical LBs, cortical LBs are more difficult to detect with routine histology (Fig. 25.1E). They tend to be found in small nonpyramidal neurons in lower cortical layers. Similar lesions in the substantia nigra are referred to as “pale bodies” (Fig. 25.1C). Ultrastructural studies of cortical LBs demonstrate poorly organized granulofilamentous structures similar to those in Lewy neurites. Not all cortical regions are equally vulnerable to cortical LBs. The frontal and temporal multimodal association and limbic cortices are most vulnerable, with the amygdala being the most vulnerable of all. It is increasingly recognized that the amygdala is also vulnerable to LBs in AD, often with little or no involvement in other areas vulnerable to LBs in PD (23,24). LBs are only rarely detected in primary cortices.
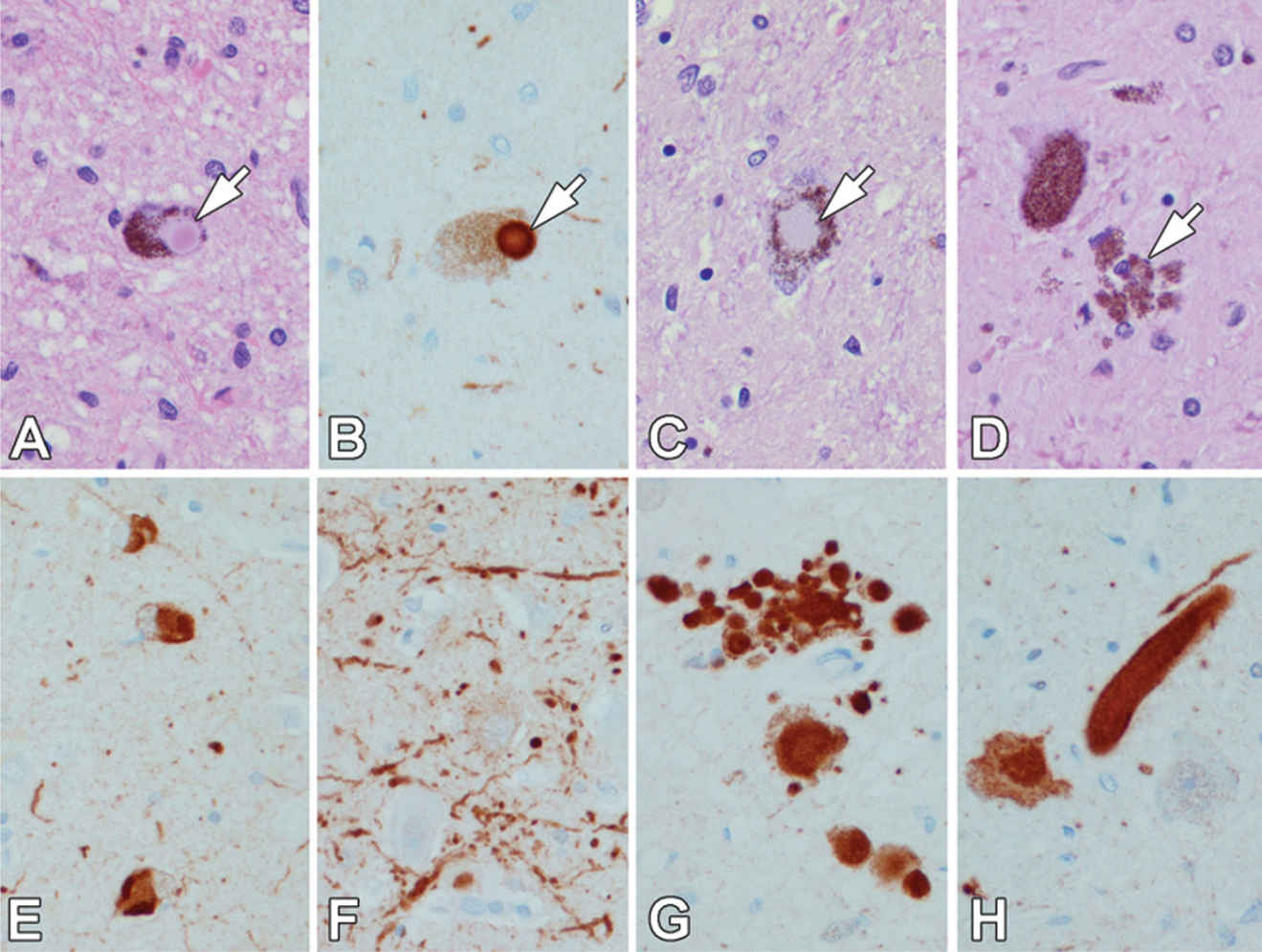
Figure 25.1. Lewy bodies (LBs) (arrow) are concentric hyaline inclusions in pigmented neurons of the substantia nigra (A). LBs are immunoreactive for synuclein (arrow in B). Pale bodies in the substantia nigra (C) are less-compact neuronal cytoplasmic inclusions (arrow). After dopaminergic neurons die, neuromelanin pigment is found in clusters of macrophages (arrow) (D). Cortical type LBs are pleomorphic inclusions in small nonpyramidal neurons in cerebral cortex (E). Lewy neurites are aggregates of synuclein in axonal processes, such as the hippocampus (F). Pleomorphic axonal (G) and large axonal inclusions (intraneuritic LBs) (H) are frequent in the dorsal motor nucleus of the vagus. (All images ×400; A, C, E – H & E stain; B, E, F, G, & H – synuclein immunostain.)
Both classical and cortical LBs share a common immunohistochemical profile. Antibodies to neurofilament were first to be shown to label LBs (25), but most neurofilament antibodies label only a subset of LBs. Ubiquitin is present in most classical and cortical LBs (13), but the most specific method of detecting LBs and Lewy neurites is synuclein immunocytochemistry (26).
Only a few biochemical studies on LBs have been reported due to the paucity of LBs in a given volume of brain tissue and their solubility properties. LBs are structurally unstable and disrupted upon biochemical purification. Initial biochemical studies suggested that a major protein constituent of LBs was a 68-kDa protein that had cross-reactivity with neurofilament protein (27). More recent studies suggest that the major structural component of LBs is synuclein (28), and in vitro studies have demonstrated that synuclein can aggregate to form filaments similar to those seen in LBs (29).
Distribution of LBs and Lewy Neurites
Neurons that are most vulnerable to LBs include the monoaminergic neurons of the substantia nigra, locus ceruleus, and raphe nuclei, as well as neurons of the dorsal motor nucleus of the vagus and cholinergic neurons in the basal forebrain and mesopontine tegmentum (30). LBs are rarely detected in the basal ganglia or thalamus, but are common in the hypothalamus, especially the posterior and lateral hypothalamus, and in the brainstem reticular formation. The oculomotor nuclear complex is also vulnerable. In the mesopontine tegmentum and the pons, the raphe nuclei and the pedunculopontine nucleus are often affected, but neurons of the pontine base are not. LBs have not been described in the cerebellum. In the spinal cord, the preganglionic sympathetic neurons of the intermediolateral cell column are most vulnerable. LBs can be found in the autonomic ganglia (both sympathetic and parasympathetic), including submucosal and myenteric ganglia of the intestinal tract (31,32). Sympathetic nerve fibers, such as those in the epicardium, are also affected (33).
Staging Synuclein Pathology in PD
Seminal studies by Braak and Del Tredici led to a proposed staging scheme of synuclein pathology in PD (34), in which the earliest stages are associated with involvement of autonomic ganglia, anterior olfactory nucleus, and the dorsal motor nucleus of the vagus. Only later in the progression of the disease does the substantia nigra become affected; symptomatic parkinsonism occurs when there has been neuronal and synaptic loss in nigrostriatal dopaminergic neurons, at which point synuclein pathology has spread well beyond the midbrain to include basal forebrain and cortical structures. This model has fueled interest in nonmotor aspects of PD and the possibility that such clinical features can be used to detect premotor or prodromal PD.
Neuropathology of PD
The brain is usually grossly unremarkable until the brain stem is sectioned; then loss of neuromelanin pigmentation in the substantia nigra and locus ceruleus becomes apparent (Fig. 25.2). The histologic correlate of pigment loss is neuronal loss in the substantia nigra pars compacta. In idiopathic PD, as in most other disorders associated with parkinsonism, the neuronal loss is most marked in the ventrolateral tier of neurons, which is known to project to the striatum. Striatal pathology is not apparent with routine histologic methods, but can be detected in immunohistochemistry for dopamine nerve termini such as with tyrosine hydroxylase immunohistochemistry (35). Neuronal loss in the substantia nigra is accompanied by astrocytosis and microglial activation. Neuromelanin pigment is often found in the cytoplasm of macrophages (Fig. 25.1D), and occasionally neurons can be detected undergoing neuronophagia, or phagocytosis by macrophages. LBs and pale bodies are found in some of the residual neurons. Similar pathology also plays out in locus ceruleus and dorsal motor nucleus of the vagus. The basal forebrain is vulnerable to neuronal loss and LBs. In PD not associated with dementia, the neocortex usually does not have many LBs, but limbic areas, particularly the amygdala, may have some. Depending on the age of the individual, varying degrees of Alzheimer-type pathology may also be detected; however, if the person is not demented, such pathology usually falls within the range expected for his or her age. Some individuals may have abundant senile plaques, but few or no NFTs, which may be a prodromal stage to AD (36).
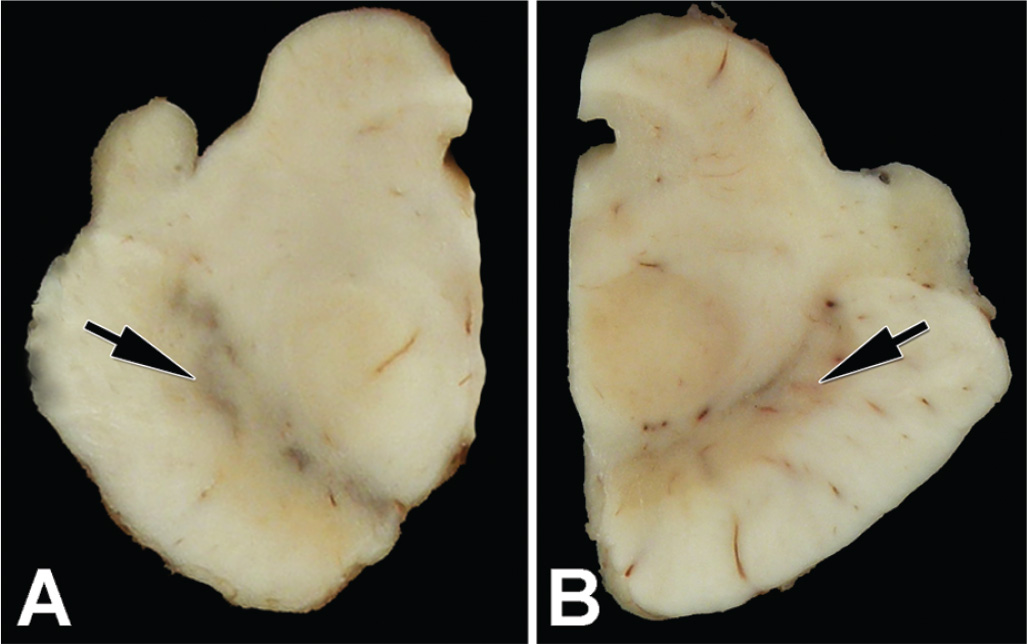
Figure 25.2. Transverse sections of hemi-midbrains of a neurologically normal person (A) and a patient with PD (B). Note the black pigment in the ventrolateral region of the substantia nigra in the normal control, but its absence in PD (arrows).
Neuropathology of Dementia in PD
Cognitive impairment and dementia is common in the course of PD (37). In the London Parkinson brain bank series, pathologic findings considered to account for dementia in PD included subcortical pathology (39%), coexistent AD (29%), and diffuse cortical LBs (26%) (38). The basal forebrain cholinergic system is the subcortical region most often implicated in dementia, and neurons in this region are damaged in both AD and LBD. Neuronal loss in the basal nucleus is consistently found in PD, especially PD with dementia (39). As previously mentioned, the basal nucleus is also vulnerable to LBs. Not surprisingly, cholinergic deficits are found in PD, PD with dementia, and DLB (40–42). Cholinergic deficits may be especially relevant to cognitive deficits in PD patients who do not have concurrent AD pathology or diffuse cortical LBs (18).
Although recent studies have shown that many PD brains have a few cortical LBs, they are usually not widespread or numerous in nondemented PD patients (18,43). When found in PD, cortical LBs are usually detected in the limbic cortices (e.g., entorhinal cortex and cingulate gyrus) and not detected in association cortices of the frontal, temporal, and parietal lobes. In contrast, several studies have shown that cortical LBs can be numerous and widespread in PD with dementia and that the density of cortical LBs correlates with severity of dementia (44,45). There are exceptional individuals who can tolerate significant numbers of cortical LBs without any apparent deficits (46). Further studies are needed to determine what factors contribute to resilience in these individuals.
Patients with widespread cortical LBs are said to have diffuse LBD. Kosaka and coworkers were the first to use the term “diffuse LBD” (47), and he subsequently subdivided diffuse LBD into pure and common forms. The latter is associated with coexistent Alzheimer-type pathology (48). Brains with cortical LBs confined to the limbic lobe are classified as “transitional LBD,” whereas those with LBs relatively limited to brain stem nuclei are said to have “brain stem LBD.” Nondemented PD subjects usually have brain stem or, occasionally, transitional LBD, whereas PD patients with dementia and patients with DLB have diffuse LBD or transitional LBD (49).
Alzheimer-Type Pathology in PD
The entorhinal cortex is susceptible to degeneration in parkinsonian disorders as well as in aging and AD. In the NFT staging scheme of Braak and Braak (50), the entorhinal cortex is among the first areas of the brain to show NFTs. In this scheme, six stages are described, with the initial stages being clinically silent. In most cases of PD with dementia and DLB, the Braak NFT stage is lower (stage IV or less) than in comparably demented subjects with AD (stage IV or greater), but usually more than in nondemented elderly normal individuals (stage III or less) (51).
Senile plaques are complicated lesions composed of extracellular deposits of amyloid and localized glial and neuronal changes (52). Given the fact that development of senile plaques is nearly inevitable with aging (53), it is not surprising that many patients with PD, and especially DLB, have variable numbers of senile plaques. This fact has become abundantly clear with the advent of molecular imaging, where amyloid is detected in most DLB and more in PD with dementia than PD without cognitive impairment (54). There are striking morphologic differences between senile plaques in PD and DLB compared with AD. In AD, senile plaques differ not only in the nature of the amyloid peptides and reactive glial changes but also in the presence of neuritic elements (i.e., neuritic plaques). In most cases of PD and DLB, the most frequent type of plaque is the so-called diffuse plaque, whereas neuritic plaques are the hallmark of AD. The importance of neuritic plaques and NFTs has been adopted by National Institute of Aging/Alzheimer Association criteria in pathologic diagnosis of AD (55).
LBs in Aging and AD
Postmortem studies of asymptomatic elderly humans demonstrate LBs, most often limited to pigmented brain stem nuclei, in up to 10% of the elderly population (56). A certain degree of parkinsonism is frequent with aging, and it is possible that the structural correlate of age-related extrapyramidal signs may be incidental brain stem LBs; however, this remains hypothetical. The pathologic substrate of extrapyramidal signs occurring late in the course of otherwise-typical AD has been the focus of only a limited number of pathologic studies. While it is commonly assumed that LBs account for extrapyramidal features in aging and AD, NFT in the substantia nigra may be more important (57,58).
MULTIPLE SYSTEM ATROPHY
The term “multiple system atrophy” is used for a neurodegenerative disease characterized by parkinsonism, cerebellar ataxia, and autonomic dysfunction, especially idiopathic orthostatic hypotension (59). There are only rare families with genetically determined MSA (60). Currently, MSA is classified into two major variants, MSA-C and MSA-P. The latter is associated with atypical parkinsonism and predominantly striatonigral degeneration, while the former has a cerebellar syndrome with olivopontocerebellar degeneration (61).
Neuropathology of MSA
The MSA brain shows varying degrees of atrophy of cerebellum, cerebellar peduncles, pons, and medulla in those with MSA-C, but prominent atrophy and discoloration of the posterolateral putamen and loss of pigment in the ventrolateral substantia nigra in those with MSA-P (Fig. 25.3). The histopathologic findings include neuronal loss, gliosis, and microvacuolation in the putamen (Fig. 25.4A), substantia nigra (Fig. 25.4B), cerebellum, olivary nucleus, pontine base, and intermediolateral cell column of the spinal cord. There is prominent brown granular pigment (in part composed of iron) in astrocytes (Fig. 25.4A) and microglia (Fig. 25.4B). White matter inevitably shows demyelination, with the brunt of the changes affecting white matter in affected areas.
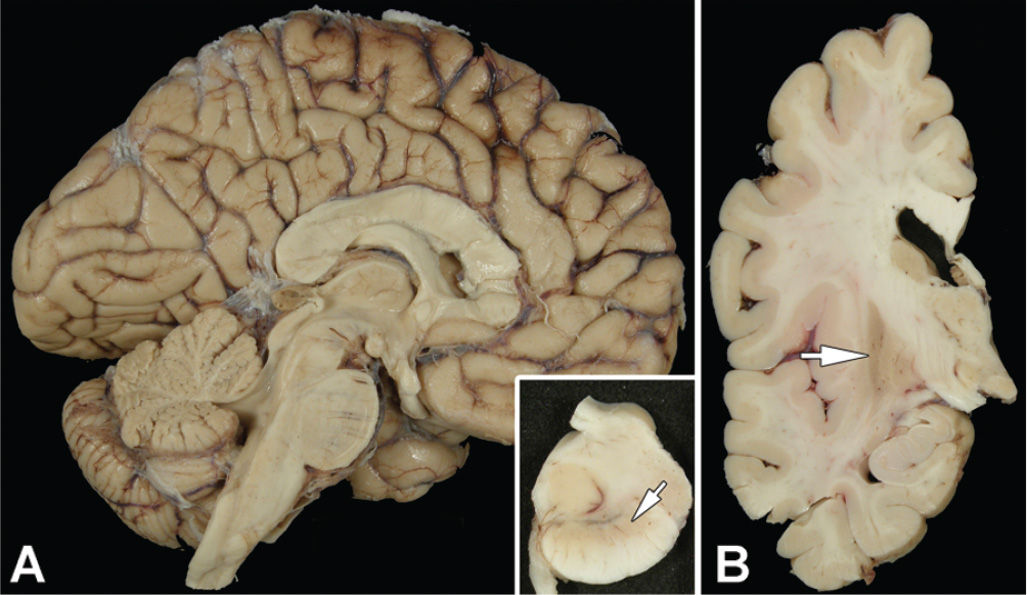
Figure 25.3. The midsagittal section of the brain of a patient with MSA-P (A) is unremarkable, except for mild dilation of the fourth ventricle. Inset in (A) shows transverse section of the midbrain with marked loss of neuromelanin pigment in the ventrolateral region (arrow). Coronal sections of the cerebral hemisphere (B) shows marked atrophy and discoloration in the lateral putamen (arrow).
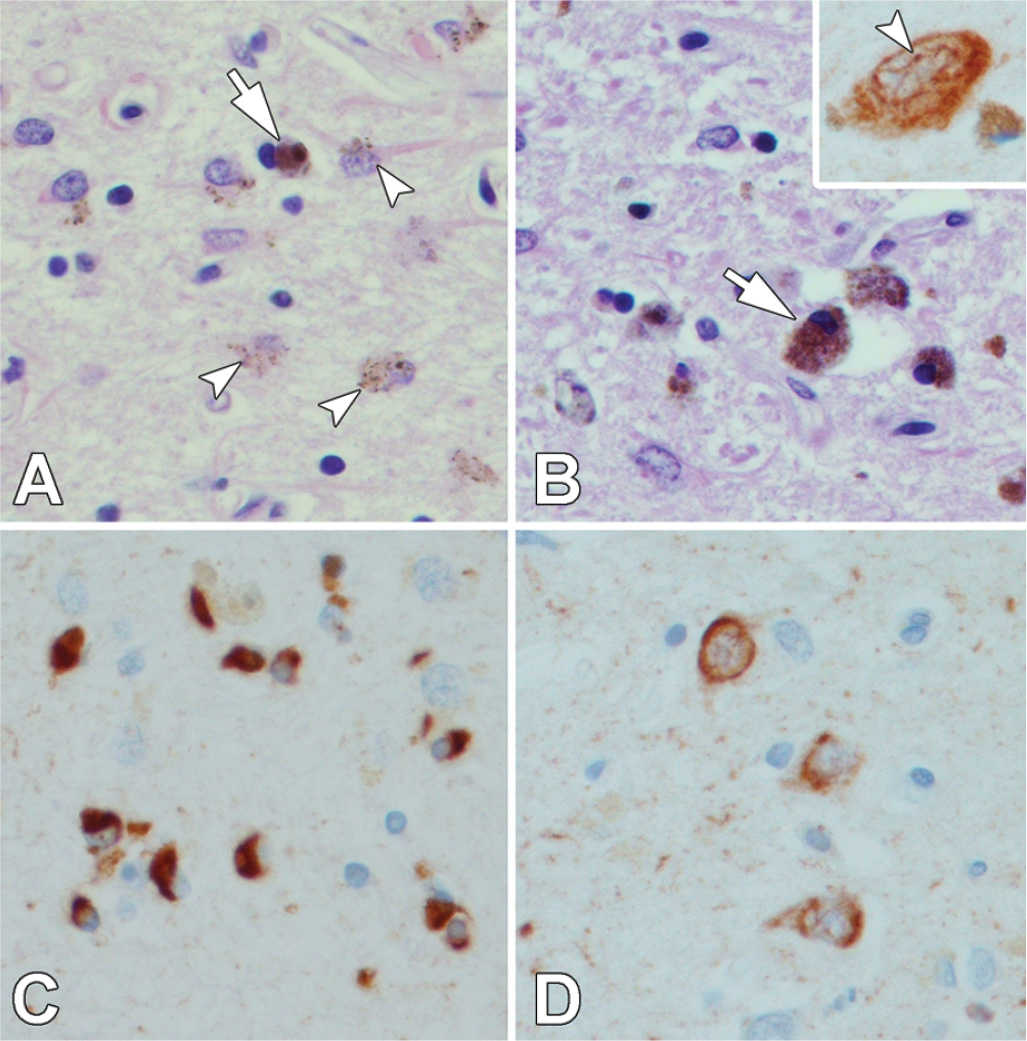
Figure 25.4. The putamen in MSA shows neuronal loss, vacuolation, and reactive astrocytosis, with accumulation of brown granular pigment (arrow), including pigment within astrocytes (arrowheads) (A). The substantia nigra has neuronal loss and pigment-laden macrophages (B). Some nigral neurons have cytoplasmic and thread-like intranuclear inclusions (B, inset). Numerous GCIs are detected in affected areas, such as putamen, with synuclein immunohistochemistry (C). Synuclein also occasionally reveals neuronal inclusions (D) and fine neuritic processes. (All images ×400; A & B – H & E stain; C & D – synuclein immunostain).
Even when the brunt of the pathology is in a striatonigral distribution, as in MSA-P, there is microscopic pathology in cerebellum, olivary nucleus, and pons. A pathologic grading scheme that takes this into account has been proposed by Jellinger (62).
Lantos and coworkers first described oligodendroglial inclusions in MSA and named them GCIs (16,63). GCIs can be detected with silver stains, such as the Gallyas stain, but are best seen with antibodies to synuclein, where they appear as flame- or sickle-shaped inclusions in oligodendrocytes (Fig. 25.4C). In addition to synuclein immunoreactivity, GCIs are immunoreactive for ubiquitin. At the ultrastructural level, GCIs are non–membrane-bound cytoplasmic inclusions composed of granule-coated, 7- to 10-nm-diameter filaments similar to those in LBs and Lewy neurites (64). GCIs are distinctly different from the oligodendroglial inclusions, so-called coiled bodies, which are found in the tauopathies. Most tau antibodies readily stain coiled bodies, but GCIs are negative. GCIs are specific for MSA and have not been found in other neurodegenerative diseases, although sparse oligodendroglial inclusions are not uncommon in the midbrain in PD (65). In addition to GCIs, synuclein-immunoreactive lesions are also detected in some neurons in MSA, but the distribution of these lesions—putamen (Fig. 25.4D), pontine base, and inferior olivary nucleus—rarely leads to difficulty differentiating them from LBs. Intranuclear inclusions, often thread-like, are detected in some neurons (Fig. 25.4B, inset). Biochemical studies of synuclein in MSA have shown changes in solubility (66) similar to changes seen in LBD.
TAUOPATHIES
Tau abnormalities play a crucial role in a group of neurodegenerative diseases known as the tauopathies. PSP and corticobasal degeneration (CBD) are the most common parkinsonian tauopathies. Other tauopathies include Pick’s disease, frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), postencephalitic parkinsonism (PEP), Guam parkinsonism–dementia complex (Guam PDC), and chronic traumatic encephalopathy (CTE) (formerly called dementia pugilistica). All tauopathies involve neurodegeneration of specific neuronal populations with filamentous tau protein aggregates in neurons and glia. Many are associated with clinical features of parkinsonism. Some are sporadic (Pick’s disease, PSP, CBD, PEP, Guam PDC, CTE), whereas others are hereditary (FTDP-17).
COMPARISON OF PSP AND CBD
PSP and CBD were first reported in the 1960s (67,68), and the term “corticobasal degeneration” was coined by Gibb and Marsden in the 1980s (69). Molecular and genetic studies have identified similar, but also distinctive changes in PSP and CBD (70,71). Furthermore, PSP and CBD share clinical and pathologic features, but notable differences warrant their current separation as clinicopathologic entities. Although individuals with CBD and PSP have bradykinesia, rigidity, and gait abnormalities, neither has a sustained clinical response to levodopa. Rest tremor, one of the cardinal clinical features of PD, is uncommon in PSP and CBD. Motor abnormalities in PSP are usually symmetric, while asymmetry is the hallmark of CBD. Focal cortical signs, such as apraxia and aphasia, are common in CBD, but not in PSP. Dementia is common in CBD and may be one of the most common presentations, but is not prominent in PSP. Supranuclear gaze palsy occurs early in most cases of PSP, but is uncommon in CBD.
Pathologically, both PSP and CBD have neuronal and glial lesions composed of tau protein. Although the anatomic distribution of lesions shows overlap, the overall pattern differs (72,73). Cortical and subcortical white matter is more affected in CBD, whereas diencephalic and brain stem white matter is more affected in PSP. The morphologies of the typical neuronal and glial lesions are distinctive (15,74).
Biochemical studies of tau protein isolated from brains of affected patients show similarities and differences in PSP and CBD. In both disorders, there is evidence of enrichment of abnormal insoluble tau isoforms (4R tau) derived from specific tau mRNA splice forms (splice forms containing exon 10) (75), but there are differences in lower-molecular-weight cleavage fragments (70). There is also evidence of similar predisposition with respect to genetic variants in MAPT (76,77). A recent genome-wide association study implicated other genes in PSP (78), but a similar study in CBD is currently lacking.
NEUROPATHOLOGY OF PSP
Gross examination of the brain often shows frontal and midbrain atrophy (Fig. 25.5A). The third ventricle and aqueduct of Sylvius may be dilated. The substantia nigra shows loss of pigment, while the locus ceruleus is better preserved. The subthalamic nucleus is smaller than expected (Fig. 25.5B). The hilus of the cerebellar dentate nucleus may be atrophic and gray due to myelinated fiber loss, and the superior cerebellar peduncle shows marked atrophy in most cases (Fig. 25.6A).
Microscopic findings include neuronal loss and fibrillary gliosis affecting multiple brain regions. The nuclei with the most marked and consistent pathology are the substantia nigra (Fig. 25.7A) and subthalamic nucleus (Fig. 25.7B), with more variable involvement of the globus pallidus. Other parts of the basal ganglia, diencephalon, and brain stem are affected to a variable degree. The cerebellar dentate nucleus has neuronal loss, grumose degeneration (Fig. 25.7C) and associated degeneration of the dentatorubrothalamic tract.
In addition to the changes visible with routine histologic methods, silver stains (e.g., Gallyas stain) or immunostaining for tau reveal NFTs and astrocytic (Fig. 25.7E) and oligodendroglial (Fig. 25.7F) lesions. NFTs in PSP often have a rounded or globose appearance (Fig. 25.7D). In addition to fully formed NFT, tau immunohistochemistry shows many pretangles, which are neurons with nonfilamentous granular cytoplasmic tau immunoreactivity that may be precursors to NFTs. Tufts of abnormal fibers, more commonly known as “tufted astrocytes” (Fig. 25.7E), are frequent in motor cortex and striatum. Tau immunohistochemistry also reveals tau-positive fibers, so-called neuropil threads, and small round glial cells in the white matter, which are oligodendroglial inclusions (“coiled bodes”) (Fig. 25.7F), especially in subcortical and brain stem fiber tracts.
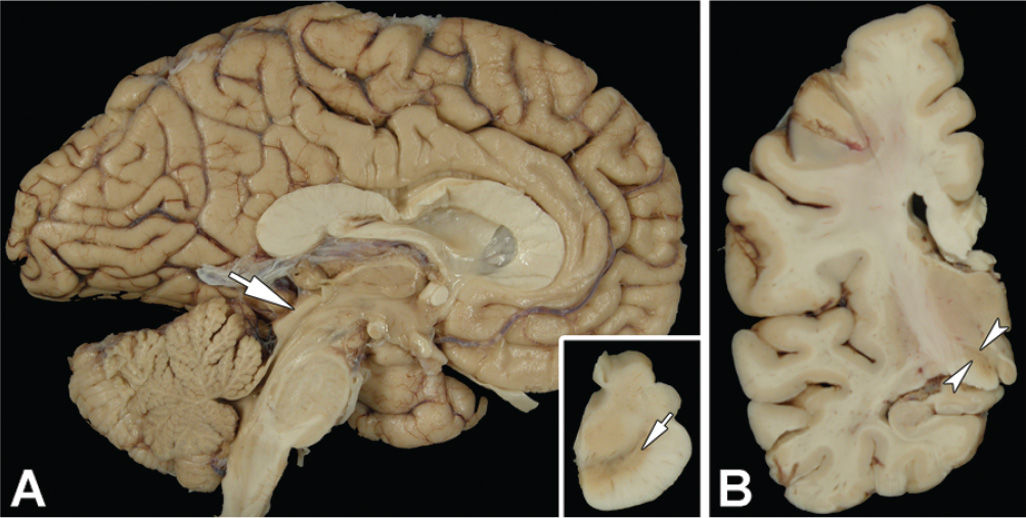
Figure 25.5. The sagittal view of the brain in PSP shows atrophy of the dorsal midbrain (A, arrow). The inset in (A) shows a transverse section of the midbrain with marked neuromelanin pigment loss, especially in the ventrolateral region (arrow). Coronal sections of the cerebrum (B) show marked atrophy of the subthalamic nucleus (arrowheads).








