Fig. 1
Invasion of (a) the choroid plexus of a lateral ventricle and (b) the leptomeninges by T. b. brucei parasites in rats infected with the parasite can occur early during the infection before invasion the brain parenchyma. Note that there is no gradient of parasites from these two areas into the brain parenchyma. Reproduced from Mulenga et al. (2001), with permission
The blood-CSF barrier is also formed by the epithelial cells of the choroid plexus, the structure responsible for CSF formation (Engelhardt and Sorokin 2009). The endothelial cells of capillaries in the choroid plexus are fenestrated and allow passage of macromolecules from the bloodstream into the choroid plexus stroma. However, the tight junctions that link the epithelial cells of the choroid plexus prevent further non-selective passage of macromolecules into the CSF (Czosnyka et al. 2004).
Other nervous system structures whose blood vessels are fenestrated and therefore are permeable to macromolecules are the peripheral nerve root ganglia and the CVOs around the third and fourth ventricles, including area postrema, median eminence, organum vasculosum of the lamina terminalis, pineal gland, posterior pituitary, and subcommissural and subfornical organs. The CVOs are enclosed by special cells called tanycytes that may restrict movement of macromolecules from the CVOs into the CSF and/or brain parenchyma.
As with leptomeninges, T. b. brucei and WBCs localize in the choroid plexus (Fig. 1a), CVOs and peripheral nerve root ganglia early after infection, before invasion of the brain parenchyma (Schultzberg et al. 1988). A particularly long slender form of the parasites, which appear mostly after three to four waves of parasitemia, has been associated with choroid plexus invasion, and this may explain why the parasites do not invade the choroid plexus during the first waves (Wolburg et al. 2012). The tight junctions between the epithelial cells of the choroid plexus may prevent further direct spread of the parasites into the CSF. Moreover, the CSF is toxic to trypanosomes (Wolburg et al. 2012) and no parasite gradient around the ventricles in the brain is observed indicating that trypanosomes do not spread through the ventricular CSF into the brain parenchyma (Masocha et al. 2004; Mulenga et al. 2001).
4.2 Invasion of the Brain Parenchyma by Trypanosomes via the BBB
The BBB, formed by cerebral endothelial cells, pericytes, basement membranes and astrocytic end-feet, is the largest and most restrictive barrier in the CNS, which prevents non-selective passage of molecules from the blood into the parenchyma (Abbott et al. 2006; Sixt et al. 2001). Tight junctions linking endothelial cells form a structural barrier, which prevents passage of macromolecules between the blood vessel and brain via the paracellular route, while the low endocytotic activity of endothelial cells, restricts passage of molecules via the transcellular route (transcytosis); this forms a functional barrier selective for specific molecules (Abbott et al. 2010; Wilhelm et al. 2011). The exchange of metabolites across the BBB mainly occurs at the level of capillaries, whereas the infiltration of WBCs during inflammation occurs at the level of post-capillary venules (Owens et al. 2008). Post-capillary venule endothelial cells are separated from the perivascular astrocytic end-feet by two basement membranes: the endothelial and the parenchymal basement membrane also called the astrocytic basement membrane (Owens et al. 2008; Sixt et al. 2001; Sorokin 2010). The composition of laminin molecules makes these two basement membranes differ with respect to WBCs penetration. The endothelial basement membranes composed of laminin α4 permit penetration of WBCs while those composed of α5 chains restrict. The parenchymal basement membranes, which have laminin α1 and α2 chains, must be “opened” before WBCs can pass (Sixt et al. 2001). Infiltrating WBCs, as occur in experimental allergic encephalomyelitis, are trapped between the endothelial and the parenchymal basement membranes before matrix metalloproteases (MMPs) are activated (Owens et al. 2008; Sorokin 2010). The focal activation of MMPs 2 and 9 in macrophages to cleave the dystroglycan receptors that anchor the astrocytic end-feet to the parenchymal basement membrane is required for further infiltration of WBCs (Agrawal et al. 2006; Sorokin 2010). Pericytes enwrapped by the endothelial basement membrane have recently been shown to be important for regulation of transcytosis across the BBB (Allt and Lawrenson 2001; Armulik et al. 2010), but their role in inflammatory processes are not yet clear.
Similar to WBCs, trypanosomes cross the BBB in a multistep process (Mulenga et al. 2001). First they cross the endothelial cell layer (Masocha et al. 2004) where capillaries are surrounded by a permissive laminin α4 containing basement membrane, followed by passage across the parenchymal basement membrane, a process regulated by different immune molecules as described further (see Sect. 4.3.2). It should be noted that brain invasion of trypanosomes and WBCs appears predominantly in the white matter, septal nuclei and diencephalic nuclei around the ventricles in experimental rodent models. This is consistent with neuropathological findings in brains from HAT victims, in which the inflammatory cell infiltration is mainly seen in the white matter. HAT is therefore considered a “leukoencephalitis”.
4.3 Molecules That Facilitate Parasite Invasion of the Brain Parenchyma
Several in vitro and in vivo studies suggest that both parasite- and host-derived factors have a role in the trypanosome invasion of the CNS (Table 1).
Table 1
Molecules/factors involved in Trypanosoma brucei spp. crossing of the BBB
Molecule/factor | Proposed roles | References |
|---|---|---|
Parasite-derived molecules | ||
Cysteine proteases (i.e. brucipain) | Increase in BBB permeability which promote parasite crossing of the BBB | |
Host derived-molecules | ||
CXCL10 | Attraction or retention of T cells and trypanosomes in the brain parenchyma | Amin et al. (2009) |
IFNα/β | Induces a limited release of CXCL10 from astrocytes and endothelial cells, which is enough for penetration of sensitized T cells and some trypanosomes into the brain parenchyma (initiation of T cell and parasite crossing the BBB into the brain parenchyma) | Amin et al. (2012) |
IFN-γ | Facilitate T cell and parasite crossing of the BBB by inducing the expression of CXCL10 and other molecules whose nature is not yet determined | Masocha et al. (2004) |
PAR2 | Brucipain interacts with PAR2 on endothelial cells to increase BBB permeability which promote parasite crossing of the BBB | |
TLR9-MyD88 signalling pathway | Induces release of the innate immune response molecules TNF-α and IFN-α/β that facilitate T cell and parasite crossing of the BBB | Amin et al. (2012) |
TNF-α | Facilitate T cell and parasite crossing of the BBB by increasing expression of adhesion molecules i.e. ICAM-1 | Amin et al. (2012) |
4.3.1 Parasite-Derived Molecules That Facilitate Trypanosome BBB Crossing
Similar to other parasite species, such as Schistosoma and T. cruzi, that secrete proteases to enable them to enter the host or the host cells, the T. b. subspecies express phosphatases and proteases on their external surfaces. Such molecules have been suggested to facilitate T. b. passage across the BBB. In vitro models of the BBB have shown that cysteine proteases play a role in parasite’s passage across cerebral endothelial cells either via the paracellular or the transcellular route (Abdulla et al. 2008; Grab et al. 2004, 2009; Nikolskaia et al. 2006a, b). The parasite-derived cysteine protease-mediated penetration of BBB depends on the activation of endothelial cell G protein-coupled receptors (i.e. protease-activated receptor-2, PAR2) and calcium signaling (Grab et al. 2009, 2011). The inhibition of parasite proteases by RNA interference resulted in prolonged survival of infected mice in one study (Abdulla et al. 2008), but whether these manipulated parasites can invade the brain was not explored. It is important to note that cerebral endothelial cells used in vitro show marked differences in the expression of molecules involved in the BBB as compared to the cells in vivo. Moreover, the structure of post-capillary venules in vitro still has to be established.
4.3.2 Host-Derived Molecules That Facilitate Trypanosome BBB Crossing
The same isolate of T. b. brucei invades the brain parenchyma of C57BL/6 mice earlier and more extensively than in SV-129/Ev mice even though the latter strain of mice higher levels of parasites in the blood (Masocha et al. 2008). Brains of T. b. brucei-infected C57BL/6 mice also express higher levels of inflammatory molecules, including pro-inflammatory cytokines and adhesion molecules than SV-129/Ev mice (Masocha et al. 2008), which suggests that genetic differences in the immune response regulation play an important role in parasite invasion of the CNS.
The role of selected immune response molecules in T. b. brucei passage across the BBB has been studied in genetically engineered mice and can be summarized as follows. The innate immune system, which recognizes pathogen-associated molecular patterns of trypanosomes via TLRs, is activated by the intracellular adaptor molecule MyD88 and plays a role in the control of parasitemia (Amin et al. 2012; Drennan et al. 2005). The induction of the cytokines TNF-α and IFN-α/β during trypanosome infection is dependent on TLR2/9-MyD88-mediated signaling (Amin et al. 2012). After T. b. brucei infection, the number of both parasites and T cells in the brain parenchyma is reduced in mice deficient in either TNF-α or IFN-α/β signaling compared to wild-type mice (Amin et al. 2012). However, T cell number is lower but parasite accumulation is increased in the brain parenchyma of TLR2 and 9 or MyD88 deficient mice compared to wild type mice (Amin et al. 2012). Thus, neuroinvasion and growth control (see Sect. 5) of the trypanosomes in the brain are regulated by different sets of molecules.
The importance of adaptive immune response molecules, and in particular T cell-derived IFN-γ, for trypanosome crossing the parenchymal basement membrane, was discovered in a series of experiments. T. b. brucei and T cell penetration of the brain parenchyma is reduced in mice deficient in IFN-γ signaling despite increased parasite levels in the blood (Masocha et al. 2004). In the absence of IFN-γ, parasites could cross the endothelium and the endothelial, but not the parenchymal, basement membrane and accumulate as cuffs in the perivascular space (Fig. 2). In accordance, IFN-γ levels are very low after T. b. brucei infection and parasite invasion of the CNS is reduced in RAG-1 deficient mice (lacking mature T and B cells) (Masocha et al. 2004). IFN-γ is secreted during the infection by antigen-specific T cells which tentatively recognize parasite epitopes on microglia or perivascular macrophages; macrophages can phagocytize parasites (Wolburg et al. 2012). IFN-γ and also IFN-α/β, albeit the latter less potently, regulate the expression of various chemokines including the T cell attractant CXCL10. Cerebral endothelial cells and astrocytes produce CXCL10 in the CNS of trypanosome infected mice (Amin et al. 2009). Mice deficient in CXCL10 or its receptor CXCR3 on T cells had less parasites in the brain parenchyma compared to wild type mice, albeit similar parasitemia levels. However the cuffing of trypanosomes and T cells around cerebral vessels observed in IFN-γ and in RAG-deficient mice was not seen in these mice (Amin et al. 2009). This suggests that different IFN-γ-regulated genes are involved in the attraction or retention of T cells and trypanosomes in the brain on one hand and in the opening of the parenchymal basement membrane for their passage on the other hand.
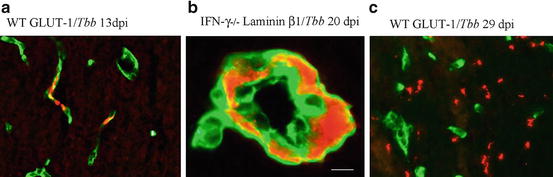
Fig. 2
Invasion of the brain parenchyma by T. b. brucei parasites (in red) in mice infected with the parasite. (a) At an early stage of the infection the parasites are confined in the lumen of cerebral blood vessels (cerebral endothelial cells in green), (b) but at a later stage they cross the endothelial cell layer and the endothelial basement membrane into the perivascular space (noticeable in the absence of IFN-γ) (basement membrane in green), (c) followed by crossing the parenchymal basement membrane to appear in the brain parenchyma (cerebral endothelial cells in green). Reproduced from Masocha et al. (2004), with permission
A scheme of cells and molecules thought to play a role in parasite and T cell neuroinvasion during trypanosome infection (Fig. 3) and a hypothetical sequence of events that may lead to invasion of predominantly the white matter (Fig. 4) can be derived from these data.
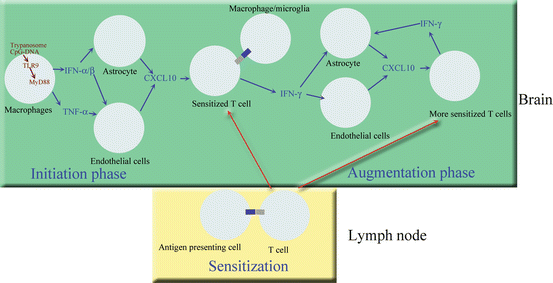
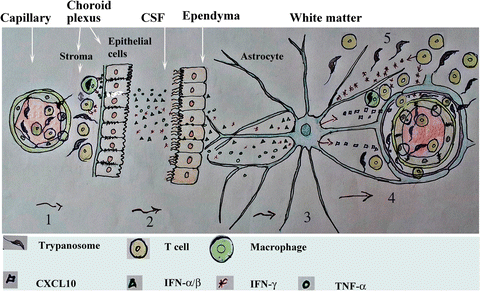
Fig. 3
Schematic diagram of the cells and molecules thought to play a role in parasite and T cell invasion of the brain parenchyma during trypanosome infection. Macrophages, after phagocytosing and lysing trypanosomes, release trypanosome CpG-DNA that activates TLR9-MyD88 signalling pathways systemically. This event induces secretion of the innate immune response molecules TNF-α and IFN-α/β. TNF-α activates endothelial cells possibly leading to expression of adhesion molecules, and IFN-α/β induces a limited expression of CXCL10 from endothelial cells and astrocytes. This could enable a few sensitized T cells and some trypanosomes to enter the brain (initiation phase). Trypanosome-derived antigens taken up and expressed by perivascular macrophages or microglia are then recognized by sensitized T cells to cause IFN-γ release. This will augment the process, since IFN-γ is a very potent inducer of CXCL10 and other molecules that lead to enhanced attraction of both T cells and trypanosomes into the brain parenchyma (augmentation phase)
Fig. 4
A hypothetical model of a series of events leading to preferential infiltration of T cells and T. b. brucei into the white matter of the brain. (1) White blood cells and parasites cross the fenestrated endothelial cell layer of capillary vessels into the choroid plexus stroma early during the infection. (2) TNF-α released by the subsequent inflammation may increase the permeability of the choroid plexus epithelial cell layer, enhancing the diffusion of cytokines into the CSF; cytokines have been found elevated in the CSF of HAT patients (Amin et al. 2009). From the ventricles, such cytokines could readily diffuse past the ependymal cells, which are not linked by tight junction, into the white matter that, in contrast to the cerebral cortex, have wide extracellular spaces allowing a rapid diffusion of macromolecules throughout. (3) IFN-α/β (and later IFN-γ) induces a limited release of CXCL10 by astrocyte and endothelial cells, (4) which permit attraction and/or retention of a few sensitized T cells and some trypanosomes into the brain parenchyma to start the initiation phase which is augmented when more IFN-γ is released as described in Fig. 3. This series of events is similar to that described for the invasion of T cells into the white matter of brain of mice infected with lymphocytic choriomeningitis virus, which is a virus that first infects cells in the choroid plexus and then microglia in the white matter (Thomsen 2009)
5 Control of Trypanosome Growth in the CNS
Patients infected by T. b. gambiense often have a long course of disease (months or several years) with signs of nervous system involvement. A recent study found that occasionally patients infected with T. b. gambiense develop trypanotolerance i.e., the infection may spontaneously cure without treatment, so that the disease is not 100 % fatal as previously thought (Jamonneau et al. 2012). Although, as mentioned above, the CSF is hostile for trypanosome survival (Wolburg et al. 2012), experimental models have suggested that trypanosomes may remain in the brain following treatment that eliminate the parasites from the rest of the body (Jennings et al. 1979). Relapses may then occur several months later, possibly from the pool of parasites in the brain. Field isolates of T. b. gambiense that cause chronic infections for 6–8 months in mice show preferential localization in the olfactory bulbs and cerebellum (Giroud et al. 2009). This suggests that growth of the trypanosomes, which in their slender form replicate rapidly, might be controlled in the brain.
It is often difficult to distinguish between the immune responses that regulate the passage of trypanosomes across the BBB from factors that control parasite replication within the brain parenchyma. As described above, we have observed that recognition of parasites via TLR2 and TLR9, but not the release of TNF-α and IFN-α/β, is required for the control of trypanosome growth in the brain (Amin et al. 2012), although the nature of the effector molecules for this are still unknown. In this context it is of interest to note that in vitro studies have shown that endogenous neuropeptides, such as vasoactive intestinal peptide (VIP), adrenomedullin, alpha-melanocyte-stimulating hormone, urocortin, corticotropin-releasing hormone and ghrelin, have potent trypanolytic activity in vitro. Administration of VIP intraperitoneally to T. b. brucei-infected mice reduces parasitemia and prolongs animal survival and interestingly, infection with T. b. brucei increases serum levels of VIP (Delgado et al. 2009). While these in vitro studies show that high levels of neuropeptides kill parasites, whether host-produced neuropeptides reach sufficiently high levels to control trypanosome growth in the CNS still requires investigation.
6 Mechanisms of Brain Dysfunctions During Trypanosome Infections
HAT is associated with symptoms both from the peripheral nervous system, e.g., pruritus and pain, and the CNS, e.g., disturbances in circadian rhythms and sleep pattern, abnormalities in movements and muscle tone, and a variety of neuropsychiatric disturbances, described in the clinical chapter (Buguet et al. 2014). The mechanisms behind these changes are not well understood, but it has been suggested that both trypanosome- and host-immune response-derived factors play roles. For instance, prostaglandin (PG) D2, which can be synthesized by both the trypanosomes and the host in response to infection, has been implicated in the pathogenesis of the pain and sleep disturbances. The immune response molecules TNF-α and IFN-γ that, as mentioned above, play roles in facilitating trypanosome crossing of the BBB, can affect synaptic functions and are involved pain and sleep pattern disruptions, similarly to some other inflammation-released molecules. Such possible mechanisms are summarized below (review, see Kristensson et al. 2010).
PGs can sensitize nociceptive neurons to painful stimuli at the peripheral and spinal cord level. Both PGE2 and PGD2 can induce hyperalgesia (Ohkubo et al. 1983), and induce or modulate allodynia (a painful response to a normally innocuous stimulus) (Eguchi et al. 1999; Telleria-Diaz et al. 2008). The cytokines TNF-α and IFN-γ can induce neuropathic pain in rodents acting possibly both peripherally and centrally (Robertson et al. 1997; Vikman et al. 2003, 2007; Zimmermann 2001). Similarly to HAT patients, hyperalgesia occurs in rats early after infection with T. b. brucei (Wiesenfeld-Hallin et al. 1991) when trypanosomes have invaded the dorsal root ganglia (Schultzberg et al. 1988), but not the brain parenchyma, and when the rats are still sensitive to drugs used for treatment of the early stage of the disease. It is therefore very important to distinguish between symptoms and signs from the peripheral and the central nervous system in clinical evaluation protocols of HAT patients, which, unfortunately, is currently not the case.
PGD2 is a potent somnogenic molecule (Huang et al. 2007; Urade and Hayaishi 2011). PGD2 receptors are located mainly in the leptomeninges covering the basal forebrain (Mizoguchi et al. 2001; Urade and Hayaishi 2011), that trypanosomes infiltrate early in infection. PGD2 is also involved in the regulation of the non-rapid eye movement (NREM) sleep (Ueno et al. 1982; Urade and Hayaishi 2011). The somnogenic effects of PGD2 are mediated through binding to its receptor causing release of adenosine that binds to adenosine A2A receptors in ventrolateral preoptic nuclei neurons (Scammell et al. 1998, 2001) and to adenosine A1 receptors in tuberomammillary nuclei neurons (Oishi et al. 2008). This promotes NREM sleep, and PGD2 synthesized by trypanosomes has been suggested to cause the sleep disturbances in HAT (Figarella et al. 2005; Kubata et al. 2000). However, HAT is characterized by disruption of the sleep rhythm, with appearance of sleep-onset-REM sleep episodes during daytime (rapid transitions from wakefulness into sleep that occurs in narcolepsy) and wakefulness episodes during night, rather than by an increase in time spent in NREM sleep, which suggests that other factors are also involved in causing the sleep disturbances in HAT.
Related posts:
 Health Problems Associated with the Use and Abuse of Khat (Catha edulis)
Health Problems Associated with the Use and Abuse of Khat (Catha edulis)
 African Trypanosomiasis: A Highly Neglected Neurological Disease
African Trypanosomiasis: A Highly Neglected Neurological Disease
 Disease: Neurology and Neurobiology
Disease: Neurology and Neurobiology
 Neurology of a Permanent and Non-progressive Motor Neuron Disorder Associated with Food (Cassava) Toxicity
Neurology of a Permanent and Non-progressive Motor Neuron Disorder Associated with Food (Cassava) Toxicity
 of Khat (Catha edulis Forsk)
of Khat (Catha edulis Forsk)
 Infections: Neurological Involvement and Neurobiology
Infections: Neurological Involvement and Neurobiology
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


