68 Other Motor Neuron Diseases and Motor Neuropathies
Clinical Presentation
Spinal Muscular Atrophy Types I–IV
Of the multiple SMA phenotypes, the infantile and childhood forms are the most prevalent. SMA type I or Werdnig–Hoffman disease is the most severe form (Fig. 68-1). Clinical manifestations become evident within the first 6 months of life. In contrast to the latter three categories, afflicted children with SMA I never develop the capability of sitting independently. In some cases, recognition of reduced movement occurs in utero or within the first few days of life. Affected infants are hypotonic with a symmetric, generalized, or proximally predominant pattern of weakness. Like ALS, facial weakness is typically mild and extraocular muscles are spared. Fasciculations are seen in the tongue but rarely in limb muscles, presumably because of the ample subcutaneous tissue of neonates. Manual tremor, so characteristic of SMA types II and III, is rarely present. Deep tendon reflexes are typically absent. Abdominal breathing, a weak cry, and a poor suck are commonplace. Ventilation difficulties stem primarily from intercostal rather than diaphragmatic weakness. Pectus excavatum and a diminished anteroposterior diameter of the chest are seen. Mild contractures may occur but arthrogryposis is not part of the classic phenotype. Intellectual development is normal. Without mechanical ventilation, death is inevitable, almost always within a year or two. An earlier age of onset correlates with a shorter life expectancy.
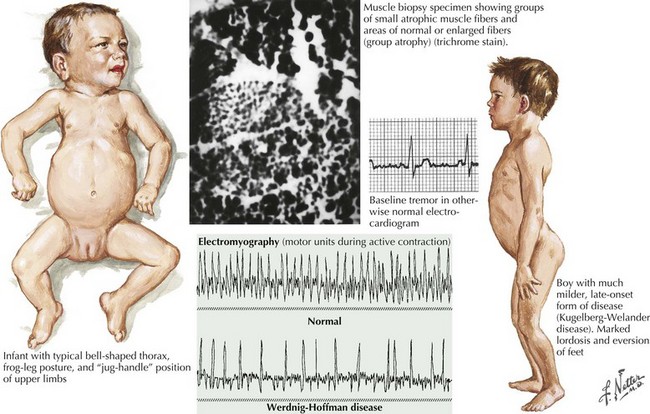
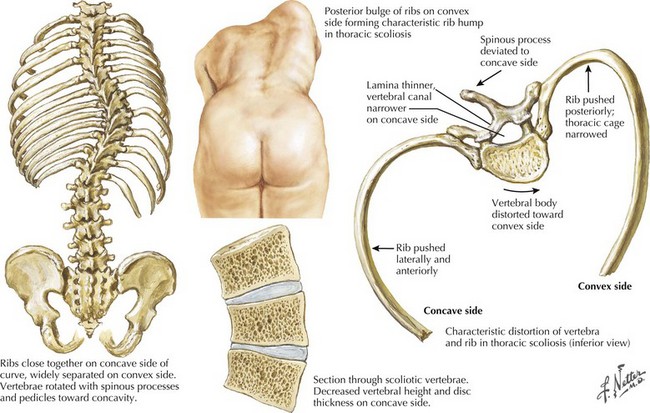
The intermediate form or SMA II typically begins between 6 and 18 months of age. The disorder is clinically defined by a child who sits independently but never walks. Postural hand tremor is the only significant phenotypic variance from Werdnig–Hoffman disease. Tongue fasciculations, areflexia, and a generalized to proximally predominant and symmetric pattern of weakness mimic the SMA I phenotype. Approximately 98% of these individuals survive to age 5 and two thirds to age 25. In view of the more protracted course and of wheelchair dependency SMA II and SMA III patients commonly acquire kyphoscoliosis and joint contractures (Fig. 68-2).
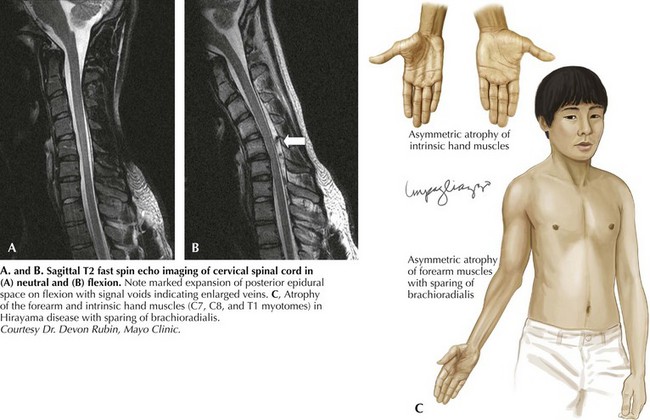
Juvenile Segmental Spinal Muscular Atrophy—Benign Focal Amyotrophy—Hirayama Disease
Hirayama disease is less frequently seen in Western populations. Ischemic changes in the cervical spinal cord of a single autopsied case of Hirayama disease led to the hypothesis of a compressive mechanism. In 2000, Hirayama reported the results of dynamic imaging in 73 patients and 20 controls. Ninety-four percent of patients had significant forward displacement and flattening of the posterior surface of the cervical cord during neck flexion (Fig. 68-3). The presumption is that the blood supply to the spinal cord is compromised, with the anterior horn representing the watershed and the most susceptible to ischemia. Other observations that supported this potential mechanism are the frequent asymmetric nature of spinal cord flattening in keeping with the asymmetric disease onset, and the lesser degree to which distortion occurred in older patients in whom progression had arrested. Nonetheless, this pathogenetic hypothesis is not universally accepted.