Specific and nonspecific pains (see Table 16.2)
Hypoaesthesia: reduced sense of touch or sensation or a partial loss of sensitivity to sensory stimuli
Hypoalgesia or hypalgesia: decreased sensitivity to painful stimuli
Hyperpathia or hyperalgesia: an excessively painful response to a mildly painful stimulus, such as a slight prick
Paraesthesia: abnormal but not unpleasant sensations, for example, tingling
Dysaesthesia: unpleasant abnormal sensations often described as a sensation of burning, pins and needles and stretching of the skin
Allodynia: a painful response to a non-painful stimulus, such as light touch
Vasoconstriction or vasodilatation
Hyperhidrosis or anhidrosis
Piloerection or loss of piloerection
Trophic changes, e.g. pale glossy skin with a sensation of coldness
Table 16.2
Pain-related symptoms in syringomyelia
Headache |
Valsalva induced |
Suboccipital |
Retro-orbital |
Generalised, nonspecific headache |
Trigeminal pain |
Orofacial pains |
Neck pain |
Segmental pain |
Radicular pain |
Back pain |
Neuropathic arthropathy pains |
Leg pain |
Scoliosis , when seen in relation to syringomyelia, presumably relates to degeneration of motor neurons innervating the spinal muscles. Once initiated, progression of the curve can occur without further motor neuron degeneration. The resulting deformity can generate discomfort of mechanical origin, in addition to the pain arising from the syrinx itself.
Following surgery for Chiari malformation, with or without syringomyelia, patients frequently enjoy a significant improvement in their quality of life (Gautschi et al. 2011; Falci et al. 2009). Headache and neck pain may diminish, as may symptoms attributable to compression of the brain stem, such as dysphagia , ataxia , nystagmus and diplopia. In contrast, symptoms directly attributable to a syrinx cavity, including pain, scoliosis, and loss of sensitivity, are the least likely to improve.
There is no clear and simple relationship between the anatomical extent of a syrinx cavity and the symptoms and signs it creates. Nor is it possible to distinguish, just by looking at their MR scans, between patients who will and those who will not develop neuropathic pain, even when this imaging is combined with electrophysiological assessments of nociceptive and non-nociceptive pathways (Hatem et al. 2010). On the other hand, higher-average daily pain intensities do correlate with greater structural damage to the spinal cord. Further, patients experiencing both spontaneous and evoked pain have less severe structural damage to the cord than do patients with spontaneous pain alone, who tend to have more severe spinal cord damage (Hatem et al. 2010).
16.3 Pain Pathophysiology and Treatment Targets
Neuropathic pain, in its various forms, is thought to result from a number of interrelated phenomena:
In general, we can describe the progression of acute pain into chronic neuropathic pain as taking place in five steps. Drugs based on different mechanisms of action can be used to target each step.
1.
Activation of Glutamate Receptors
Glutamate transmitter release results in increased activation of spinal receptors and increased neuronal excitability. Release of the glutamate is calcium channel dependent. Analgesics such as gabapentin and pregabalin target these altered calcium channels and inhibit their function.
2.
Activation of the N–methyl–D–aspartate (NMDA) Receptor
In the spinal cord, release of peptides and glutamate activates the NMDA receptor , which, in concert with other spinal systems, generates a persistent pain state. Wind-up 8 and long-term potentiation 9 are key processes related to chronic activation of NMDA receptors. Wind-up is induced by C-fibre and A-delta fibre inputs and, once produced, enhances all responses, including those from low-threshold inputs. If the peripheral sensory input declines, there might be a slow return of neuronal responses back to baseline, so blocking such peripheral drives should attenuate central sensitisation. Unfortunately, in some cases chronic pain does not cease, most probably because of glial activation (see below). Long-term potentiation is a longer-lasting version of wind-up, where high-frequency C-fibre input produces chronic excitability, an event that persists even though the input is terminated. Ketamine blocks the NMDA receptor complex, and use of NMDA antagonists has been a useful tool for demonstration of NMDA receptor-mediated hypersensitivity in patients with neuropathic and complex regional pain syndrome pains (Azari et al. 2010).
3.
Temporal Summation (Wind–up and Further Wind–Up)
If the nociceptive input continues, neuronal responses remain elevated, resulting in a cascade of detrimental neuronal overactivity. By this process weak stimuli may evoke pain, if repeated or if their duration is prolonged.
4.
Glial Activation (See Below)
5.
Cortical Reorganisation (See Below)
Spinal cord neurons that become hyperexcitable, as a result of the mechanisms described above, show reduced thresholds to normal sensory inputs, greater evoked responses to such input, increased receptive field sizes10 and ongoing stimulus-independent activity. These processes are all important factors in the pathogenesis of allodynia, hyperalgesia and spontaneous pain. Overactive neurons in the central nervous system can be inhibited via drugs that target neural cells directly such as antidepressants, antiepileptics, GABAergic agonists (benzodiazepines) and opioids . A common feature of these drugs is that their targets are ion channels and receptors on nerve endings (synapses).
16.4 The Role of Glial Cells in Neuropathic Pain
Virchow (1821–1902) first described and depicted glia as gelatinous material giving structural support to the nerve cells. In 1894 Franz Nissl described the morphological changes seen in glial cells following spinal cord injury, regarding these as a biological response to promote nerve repair. These days we are increasingly aware of other roles played by the glial cells, including in the development of neuropathic pain. Until recently development of new analgesics and treatment of neuropathic pain has focused on neuronal targets. The vital role played by glial and inflammatory cells has been overlooked but is now a fast-emerging area of research (Bulanova and Bulfone-Paus 2010). A concept, which the author refers to as ‘the hexapartite synapse’ , describes six interconnecting elements that play a functional role in neurotransmission of pain (Fig. 16.1). The hexapartite synapse consists of two neurons making synaptic contact, a microglial cell, an astrocyte , a T-cell lymphocyte and a mast cell . All these neuronal and non-neuronal cells function as a unit, and non-neuronal cells can influence the generation of electric impulses. The familiar paradigm of one afferent and one efferent neuron with a synapse in between is obsolete, and consequently therapies that only target the neuron are likely to be inadequate (Fields 2009; Keppel Hesselink 2011).
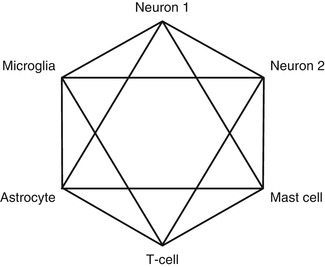
Fig. 16.1
The hexapartite synapse. Six cellular elements play a functional role in the genesis and maintenance of neuropathic pain: two neurons, a microglia cell, an astrocyte , a mast cell and a T lymphocyte. The non-neuronal cells play an underestimated role in neuropathic pain, and failure to deal with this is one of the major reasons for unsatisfactory control of neuropathic pain
Damage to the sciatic nerve in rats causes astrocytes in the dorsal horn of the spinal cord to increase in volume and to multiply, a pivotal factor in the development of neuropathic pain (Garrison et al. 1991). Low-grade inflammation also develops in the spinal cord dorsal horn and along the pain pathways to the thalamus, as well as further upstream, as far as the parietal cortex (Saade and Jabbur 2008). A consequence of this neuro-inflammation is glial cell activation, especially of the microglia (Aldskogius 2011; Gao and Ji 2010a). This activation also takes place in central pain (Wasserman and Koeberle 2009). Activated microglia and astrocytes then release many irritant molecules such as proinflammatory cytokines , including interleukins , chemokines and tumour necrosis factor (TNF)-alpha, which contribute to chronic pain states. Hyperactive neurons produce comparable compounds, such as growth factors which in turn activate spinal cord microglia and astrocytes, and a vicious circle emerges, where both cell types wind up each other and neuropathic pain is both initiated and maintained (Graeber 2010). In the rat spinal cord, astrocytes are responsive to the pain neurotransmitter substance P (Marriott et al. 1991). There is an intimate interaction between neuronal and non-neuronal cells. For example, following nerve cell injury, certain enzymes are activated, such as c-Jun N-terminal kinase in spinal cord astrocytes, leading to the expression and release of monocyte chemotactic protein-1 (MCP-1). Monocyte chemotactic protein-1 is a cytokine which increases pain sensitivity via direct activation of NMDA receptors in the spinal cord. c-Jun N-terminal kinase plays a key role in the body’s response to stressful stimuli such as inflammatory signals and changes in levels of reactive oxygen species. c-Jun N-terminal kinase activity regulates several important cellular functions, including cell growth, differentiation, survival and apoptosis, and pharmacological inhibition of c-Jun N-terminal kinase attenuates neuropathic pain in animal models (Wang et al. 2011). In addition to spinal wind-up phenomena like these, cortical reorganisation adds to the complexity of the central sensitisation processes (Dimcevski et al. 2007). The term cortical reorganisation refers to the functional changes that occur in parts of the brain. An extensive network of brain regions often referred to as the ‘pain matrix ’ frequently show abnormalities on functional imaging studies in chronic pain states, and changes in the motor and sensory homunculus have also been described (Henry et al. 2011).
There are currently five major neurobiological pathways known to activate microglia (Fig. 16.2) (Smith 2010).
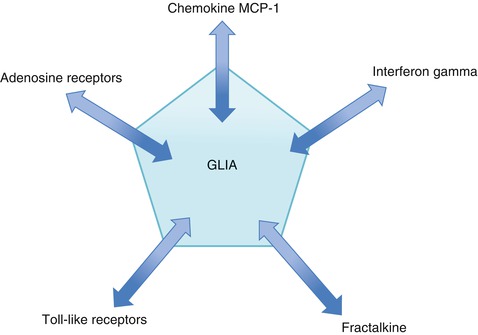
Fig. 16.2
Microglia-activating pathways. There are many targets to inhibit overactive glia in neuropathic pain
1.
Fractalkine (also known as chemokine (C-X3-C motif) ligand 1 or neurotactin) is a chemokine11 produced by a variety of cells, including neuronal and glial cells . It induces microglia chemotaxis . Microglia are the only central nervous system (CNS) cells that express the fractalkine receptors, which are upregulated in pain states. Upon activation, microglia secrete proinflammatory mediators such as prostaglandins, proteases, cytokines (TNF-alpha, interleukin-1 beta, interleukin -6) and excitatory amino acids , whose receptors are expressed on dorsal horn neurons. It is speculated that it is by this process that microglia alter sensory neuronal activity (Owolabi and Saab 2006).
2.
Interferon gamma (INF-γ) is a cytokine with antiviral, immunoregulatory and antitumour properties. Interferon gamma alters transcription in up to 30 genes producing a variety of physiological and cellular responses. After becoming activated with INF-γ, microglia release further interferon gamma which activates more microglia and initiates a cytokine-induced activation cascade, rapidly activating all nearby microglia.
3.
Monocyte chemotactic protein–1 (MCP-1) is also known as chemokine (C-C motif) ligand 2 (CCL2) or small inducible cytokine A2. Monocyte chemotactic protein induces monocyte, macrophage, basophil and mast cell migration and is synthesised by monocytes, macrophages, dendritic cells and astrocytes (Deshmane et al. 2009). This cytokine contributes to the pathogenesis of monocyte-dependent tissue injury, and release is triggered by increasing NMDA concentrations (Szaflarski et al. 1998). The consequence of increased MCP-1 concentrations and upregulation of the chemokine receptor CCR2 and MCP-1/CCL2 is an enhanced and prolonged persistent pain state (White and Wilson 2008; Gao and Ji 2010b).
4.
Toll–like receptors (TLR) are membrane-spanning receptors that have a pivotal role in the innate immune response12 in particular cellular activation and cytokine production in response to microbes. In the CNS TLR4 is expressed exclusively by the microglia, and TLR4 mRNA expression is significantly increased in experimental neuropathic pain states (Smith 2010) (Fig. 16.3).
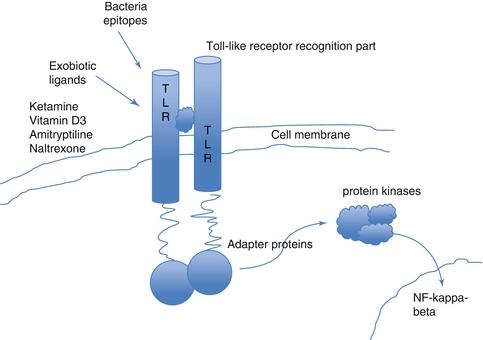
Fig. 16.3
Toll-like receptors. The Toll-like receptors are membrane-spanning receptors (named TLR1 to TLR13). The receptors function as dimers, and after activation, for instance, by an analgesic drug, Toll-like receptors recruit adapter molecules within the cytoplasm of cells to propagate a signal leading to the induction in the nucleus of certain key genes or the suppression of other genes that orchestrate the inflammatory response and chronic pain states. Toll-like receptors can be found on mast cells, glia and many immune-competent cells as well as on neurons. These receptors might play an important role in glia modulation by drugs such as ketamine, propofol, vitamin D3 or low-dose opiate antagonists such as naltrexone
5.
P2X receptors (receptors for the nucleotide adenosine). Nucleotides that are released and leaked from cells are involved in cell-to-cell communication in physiological and pathophysiological conditions (Smith 2010). The upregulation of P2X4 receptor in microglia appears to be an important process in contributing to neuropathic pain (Smith 2010).
It is increasingly recognised that these phenomena play a central role in neuropathic pain. Gliopathic or asteropathic pain may become new synonyms for neuropathic pain, and glia-modulating drugs may become a new class of neuropathic pain drugs (Ohara et al. 2009). Other non-neuronal targets, for instance, gap junctions and connections,13 are still in an early phase of research and development. These new non-neuronal targets will also lead, hopefully, to additional avenues of pain medication (Wu et al. 2012).
16.5 Traditional Pharmacological Therapies
Most outcome studies looking at the treatment of neuropathic pain are focused on painful peripheral polyneuropathies, especially diabetic and postherpetic neuralgias. Studies of central neuropathic pain are few in number, largely because it is laborious to recruit sufficient patients for entering into clinical trials. There is therefore little data on nonsurgical management of neuropathic pain secondary to Chiari malformation and syringomyelia and certainly no methodologically sound outcome studies. The literature is confined to anecdotal recommendations, case reports and series with small patient numbers. In addition, the majority of the animal models used in drug trials are not representative of the real-life situation for human patients. The only comparable animal models relevant to syringomyelia are some breeds of toy dog , especially Cavalier King Charles Spaniels (see Chap. 14), which have a high prevalence of syringomyelia (Knowler et al. 2011). The opportunities that this model may represent have not yet been realised, with only a few unpublished clinical trials having taken place.
Drawing conclusions about what is the most effective therapy is even more difficult when one has to take into account the many differences in study design (Table 16.3).
Table 16.3
Variations in study design between different trials
Population differences |
Race |
Sex |
Age |
Patient numbers |
Power calculations |
Estimated magnitude of effect |
Number of subjects included |
Number of subjects dropping out |
Trial duration |
Inclusion and exclusion criteria |
Pain intensity |
Outcome measures |
Whilst our understanding of neuropathic pain-generating mechanisms has grown considerably, this has not been translated into a similar improvement in treatment efficacy, and most neuropathic pain patients are still left with insufficient pain relief (Finnerup et al. 2010). Clear insights into why some patients are nonresponders remain absent. Given the high variability in intensity, severity and location of symptoms, each patient must receive an individualised treatment plan.
The analgesics most often used for treatment of spinal cord injury -related pain are nonsteroidal anti-inflammatory drugs (NSAIDs), acetaminophen (paracetamol ) and non-opioid muscle relaxants, such as baclofen and tizanidine . Generally, these analgesics are not prescribed by pain physicians but are purchased by patients themselves or prescribed by general practitioners. As single agents, or in combination, they are, unfortunately, ineffective in many spinal cord injury patients (Cardenas and Jensen 2006). They also have potential risks due to gastrointestinal, renal and hepatic toxicity, especially with prolonged use and higher dosage.
From the perspective of most pain specialists, antidepressants, antiepileptics and opioids are the best established and most commonly used adjuvant analgesics. Most have similar equivalent efficacy, with a number needed to treat14 (NNT) of 3–6, but the adverse effect profiles differ (Finnerup et al. 2010).
16.5.1 Tricyclic Antidepressants
Tricyclic antidepressants such as amitriptyline and nortriptyline have ‘dirty’15 pharmacology, which is perhaps the reason for their efficacy in pain states with complex pathophysiology. Pharmacologically dirty drugs, which bind to multiple receptors, tend to be more effective for neuropathic pain but have more potential adverse effects. The therapeutic effect of the classical tricyclic antidepressants is mediated by their inhibition of the reuptake of noradrenaline and of serotonin. However, they also interact with the muscarinic acetylcholine receptor, the histamine-1 receptor, the alpha-1 adrenergic receptor and sodium ion channels (Pancrazio et al. 1998). The more receptors that are triggered, then the greater the biological effect. If only one receptor in a complicated network is influenced, then this impact will ultimately be neutralised. If multiple sites are affected, then the network is more likely to be broken and for longer. There have been some anecdotal reports that tricyclic antidepressants are effective for neuropathic pain following spinal cord injury in the presence of depressive symptoms and less effective in patients not suffering from signs of depression (Attal et al. 2009; Rintala et al. 2007).
Tricyclic antidepressants are mostly metabolised by cytochrome P450 2D6 (CYP2D6); therefore drug interactions and high serum concentrations can occur, as this hepatic enzyme plays an important role in the metabolism of many different drugs and compounds. The same dose can result in 10- to 30-fold variation in serum concentration between different patients. Patients who metabolise the drug slowly may sometimes develop a toxic serum concentration following just a single oral dose or, rarely, even after topical application. At the same time, even a small amount of amitriptyline can have a positive effect. For example, 10 mg amitriptyline before bedtime may improve sleep and decrease nocturnal pains.
16.5.2 Serotonin Antagonist and Reuptake Inhibitors
Serotonin antagonist and reuptake inhibitors are a class of drugs, most commonly prescribed for depression, which act by antagonising serotonin receptors and/or inhibiting the reuptake of serotonin, norepinephrine or dopamine. Additionally, most also act as alpha1-adrenergicreceptor antagonists. Examples of this class of drugs include venlafaxine , duloxetine and trazodone . Compared to tricyclic antidepressants, SARIs have a more selective action, and as they are cleaner in their receptor affinities, there are fewer histaminergic and muscarinergic adverse effects. Whether this theoretical advantage translates into better tolerability for neuropathic pain has not been substantiated in comparison trials. Compared to amitriptyline, serotonin antagonist and reuptake inhibitors have a higher NNT and are therefore less efficacious for neuropathic pain. A recent trial compared duloxetine and pregabalin for patients with diabetic peripheral neuropathic pain, who had inadequate pain control following gabapentin monotherapy. Duloxetine was not superior to pregabalin as a monotherapy, and there was no synergistic effect with combination therapy . On the contrary, the efficacy of the combination was less than the efficacy of duloxetine alone. In addition, adverse effects such as nausea, insomnia, hyperhidrosis and decreased appetite were more common with duloxetine than pregabalin (Tanenberg et al. 2011).
16.5.3 Antiepileptic Drugs
Carbamazepine seems to be effective for peripheral neuropathic pain but has not been subjected to a clinical trial lasting longer than 4 weeks (Wiffen et al. 2011b). In patients with central, post-stroke pain, there was no difference in efficacy between amitriptyline and carbamazepine (Selph et al. 2011).
Gabapentin and pregabalin target the alpha-2-delta subunit of the voltage-dependent calcium channel. The efficacy of both is comparable, but due to the higher affinity of pregabalin for the receptor, a lower dose is required to achieve optimal analgesia. Gabapentinoids have been evaluated specifically in spinal cord injury pain, with positive effects (Vranken et al. 2008; Tai et al. 2002; Levendoglu et al. 2004). A Cochrane review investigating gabapentin in randomised trials for acute, chronic or cancer pain concluded that gabapentin was superior to placebo in 14 of 29 studies (Moore et al. 2011). Patients taking gabapentin can expect to have at least one adverse event (66 %), and some will withdraw because of such effects (12 %). Common side effects include dizziness (21 %), somnolence (16 %), peripheral oedema (8 %) and gait disturbance (9 %). To date, no head-to-head comparison studies are available for clinicians to determine whether one of these drugs is superior for central neuropathic pain (Tzellos et al. 2008). These studies are not popular to conduct and are very rarely, if ever, sponsored by the manufacturer.
Benzodiazepines may inhibit some of the ectopic activity16 in peripheral nerves following nerve injury and consequently may be used in the management of neuropathic pain, but efficacy has never been proven in well-controlled trials (Reddy and Patt 1994). There are a few old reports, from small, open-label trials,17 where clonazepam was successfully used to treat burning mouth syndrome and trigeminal neuralgia (Smirne and Scarlato 1977; Court and Kase 1976). It is not uncommon for clonazepam to be used for spinal cord injury pain, but supportive data is lacking. Doses of up to 8 mg of clonazepam often result in marked drowsiness, so, typically, a lower dose of up to 2 mg is used. Although not supported by enough data, sometimes one may want to explore the usefulness of clonazepam in central neuropathic pain, especially in case of muscle spasms.
Lamotrigine initially appeared promising in animal models of peripheral neuropathic pain and perhaps more effective than compounds such as carbamazepine and gabapentin (Chogtu et al. 2011). Clinical trials have not, however, been convincing, and there is no solid evidence supporting the use of this drug in neuropathic pain (Selph et al. 2011).
A number of antiepileptic drugs have been investigated and have shown no efficacy for pain management in patients after spinal cord injury. These include valproic acid and its sodium salt (Drewes et al. 1994; Gill et al. 2011), levetiracetam (Finnerup et al. 2009) and lamotrigine (Wiffen et al. 2011a). Compounds such as phenytoin , topiramate and carbamazepine have not been studied in post spinal cord injury pain.
For spinal cord pain and post-spinal cord injury neuropathic pain, there is enough evidence to support the use of gabapentin and pregabalin (Ahn et al. 2003; Siddall et al. 2006; Putzke et al. 2002). Evidence to support the use of other antiepileptic drugs is less substantial, and results of long-term studies have not yet been published (Eisenberg et al. 2007).
16.5.4 Opioids
Opioids have been used for pain management for thousands of years and are mentioned in pivotal medical texts of the ancient world. They were used extensively for chronic pain management and palliative care in the nineteenth and early twentieth century. In the 1970s and 1980s, opioids were not considered useful for the management of neuropathic pain, and one definition of neuropathic pain was that which was unresponsive to opioids (Arner and Meyerson 1988). This view, which was founded on results from small, mostly short duration and uncontrolled trials, is no longer held. Even so, trials of opioid use in central neuropathic pain are still scarce. Tramadol and tapentadol have been evaluated in peripheral neuropathic pain conditions, but the efficacy in central neuropathic pain states is not widely tested. One relatively small but randomised trial does, however, support their use in post-spinal cord injury pain (Norrbrink and Lundeberg 2009).
A study comparing methadone to placebo, for neuropathic pain, demonstrated evidence of analgesic effect at a dose of 20 mg/day but not at a dose of 10 mg/day (Cherny 2011). Methadone does have several distinct advantages over other opioids, particularly as it has no active metabolites. Classic opioids like morphine , oxycodone and fentanyl are broken down into metabolites that are ‘hyperalgesic’, that is, molecules that can cause pain when they accumulate under conditions of chronic administration. This means that patients taking opioids may experience more pain or even allodynia , a phenomenon referred to as opioid-induced hyperalgesia Consequently, methadone is a good alternative when intolerable adverse effects from another opioid limit further dose escalation. Often a much lower dose is required than would be expected from equianalgesic conversion tables. In addition, methadone is comparatively inexpensive, and with chronic use, the long duration of analgesia allows less-frequent dosing than is required with other opioids. Methadone is therefore often regarded as a logical choice for controlling malignant and non-malignant chronic pain (Portenoy and Foley 1986) although this view is not universally held, partly because of the risk of fatal overdose. Some recommend that methadone should not be the first-choice drug for pain and nor should it be used in opioid-naive patients (Terpening and Johnson 2007). Nor is there a clear consensus on the appropriate interval for dosing of methadone with recommended intervals ranging from 3 to 24 h (Ripamonti et al. 1997). The duration of analgesia following a single dose of methadone is 4–6 h. One study in which patients controlled their own dosing interval, at a fixed 10 mg dose, showed that after a week of repeated dosing, the initial 3- to 7-h interval lengthened to an average of 10 h (Sawe 1986). The recommended starting dose in an opioid-naive patient is 2.5 mg orally every 8 h. Frail elderly patients may require a lower initial dose, and 2.5 mg orally, once daily, has been suggested (Toombs and Kral 2005).
There is some limited data supporting intravenous use of opioids (oxycodone) for spinal cord injury patients with anticonvulsant refractory neuropathic pain (Barrera-Chacon et al. 2011). A 3-month follow-up prospective, multicentre study, following 54 patients, concluded that oxycodone, in combination with anticonvulsants, decreased pain intensity and diminished the impact of pain on physical activity and sleep. However, half of the patients showed at least one treatment-related adverse event, with constipation being the most frequent, in one-third of patients.
16.5.5 NMDA Receptor Antagonists
Ketamine is an anaesthetic drug and an N-methyl-D-aspartate (NMDA) receptor antagonist. It also inhibits pain by a number of other routes, including depression of the Toll-like receptor 3 pathway, via certain ion channels and downregulating activated glia (Hayashi et al. 2011; Mei et al. 2011a, b). Ketamine is an old drug which has been in clinical practice for nearly 40 years and may have a valuable role in management of refractory neuropathic pain patients (Cohen et al. 2011; Truin et al. 2011; Zhou et al. 2011). The effective dose for analgesia is much lower than that required for anaesthesia. This is thought to be due to a second mechanism of action, most likely via the Toll-like receptors on the glia (Mei et al. 2011b).
The usefulness of ketamine, delivered via a patient-controlled intravenous delivery system, has been reported in a single patient with cervical syringomyelia (Cohen and DeJesus 2004). The regime used resulted in a significant lessening of the pain, permitting a reduction in opioid dosage (Cohen and DeJesus 2004). It has also been reported that relative short courses (4 days) of intravenous ketamine infusion could trigger long periods (11 weeks) of decreased pain in complex regional pain syndrome type I (Sigtermans et al. 2009). The intravenous preparation can also be given orally, sublingually or rectally or as a spray for intranasal delivery.
16.5.6 Other Agents
Intravenous lidocaine infusion can be used for treating post-spinal cord injury pain in the short term but is not an option for chronic therapy (Attal et al. 2000). In syringomyelia, spasticity contributes to a patient’s discomfort, and spasmolytics such as baclofen and tizanidine can be useful co-analgesics (Devulder et al. 2002). Intrathecal baclofen is also useful for reducing pain and spasticity after spinal cord injury (Lind et al. 2004).
Pharmaceutical trials of drug therapy for hyperhidrosis caused by spinal cord lesions are few and are not supportive for their given therapies, for example, dextropropoxyphene hydrochloride (Andersen et al. 1992).
16.6 Novel Drug Therapies
16.6.1 Cannabis and Endocannabinoids
Cannabinoids such as tetrahydrocannabinol, cannabidiol and nabilone can be useful in several pain states including central and peripheral neuropathic pain, rheumatoid arthritis and fibromyalgia (Lynch and Campbell 2011). Smoked cannabis is a method used by a number of individuals with chronic, noncancer pain as well as by some patients with multiple sclerosis. Many natural and synthetic cannabinoids are therefore under investigation as potential anti-neuropathic pain drugs (Rahn and Hohmann 2009). Despite several papers supporting the efficacy and safety of cannabis and cannabinoids for various pain states, the medical use of cannabis is forbidden in many countries.
Palmitoylethanolamide is an endogenous fatty acid amide which can be found in tissues of all mammals, including man, and some foods, such as eggs and milk (Costa et al. 2002). It functions as a ubiquitous signalling molecule and is formed in the brain from the membrane phospholipid N-acylated phosphatidylethanolamines (Hansen 2010). It mimics several endocannabinoid-driven actions, even though it does not bind to cannabinoid receptors 1 and 2 (Scuderi et al. 2012). Palmitoylethanolamide can activate many receptors, most notably the peroxisome proliferator-activated receptors. These are cell nuclear receptors, mediating several physiological functions including lipid metabolism, energy balance and inflammation. There have been many scientific studies detailing palmitoylethanolamide’s neuroprotective, antiepileptic, anti-inflammatory and analgesic properties (Lo Verme et al. 2005; Koch et al. 2011; Esposito et al. 2011; Loria et al. 2008; Gatti et al. 2012). Palmitoylethanolamide also downregulates hyperactive mast cells and is a possible candidate for treating several chronic inflammatory diseases (Aloe et al. 1993) including neuroinflammatory disorders (Skaper and Facci 2012). Palmitoylethanolamide is available for clinical use for the treatment of chronic pain and chronic inflammation in some parts of Europe. A suggested dosing regimen for chronic pain is 600 mg palmitoylethanolamide twice daily for 3 weeks followed by single daily dosing in addition to standard analgesic therapies or as single therapy (Gatti et al. 2012). To date no drug-to-drug interactions have been documented (Gatti et al. 2012).
Other endocannabinoids include oleoylethanolamide, stearoylethanolamide, 2-lineoylglycerol, 2-palmitoylglycerol and anandamide or arachidonoylethanolamide. Many of these have anti-inflammatory and/or analgesic actions and have been investigated for potential therapeutic benefit (LoVerme et al. 2006; Calignano et al. 2001; Costa et al. 2008; Conigliaro et al. 2011; Indraccolo and Barbieri 2010).
16.6.2 Naltrexone
Experiments in animals have demonstrated that a transient blockade of opioid receptors, by low doses of an antagonist such as naltrexone, can stimulate increased production or upregulation of mu-opioid receptors in pain centres in the brain (Mannelli et al. 2006). Low doses of opioid antagonists have therefore been postulated to ‘reset’ the opioid-receptor system for a period of time. In addition, low-dose naltrexone also inhibits glial cell activation, via Toll-like receptor 4, which might have an analgesic effect (Inceoglu et al. 2006; Mattioli et al. 2010). It has been suggested that naltrexone has two dose-related effects: at a low dose of 1–5 mg, the Toll-like receptors are targeted and opioid receptors are reset, and at 10 mg and above, opiate receptors are blocked. A low dose of an antagonist transiently blocks the opioid receptors, resulting in increased production, or upregulation, of mu-opioid receptors in regions of the brain that control pain responses. After the antagonist effects wear off (depending on the agent and dose, this may take minutes to hours), then there are increased numbers of receptors able to bind endogenous or exogenous opioids. In addition, the body responds to the temporary opioid-receptor blockade by increasing production of endorphins. This upregulation of the opioid receptions can also ‘reset’ the opioid receptors from the desensitisation which occurs during chronic opioid treatment. However, practical use of naltrexone is still experimental and requires experience in pain management, in particular, and knowledge of the half-life of the opioids that the patient is already receiving. Typically low-dose naloxone is not combined with opioids, and patients are advised to take 1.5–4.5 mg at bedtime. Occasionally patients do report adverse effects, such as vivid dreams, nightmares or night-time waking. For these patients a morning prescription of low-dose naltrexone can be taken. Although this is contrary to normal practice, we have not found any sound scientific data indicating that low-dose naltrexone should not be administered in the morning (Leavitt 2009).
16.6.3 Magnesium
Magnesium is a physiological blocker of the NMDA receptor , and it has been suggested that magnesium supplementation may have an anti-nociceptive effect. Although initial studies looked promising (Lee et al. 2012), their findings were not supported in a recent clinical trial (Pickering et al. 2011). This trial, however, had a high placebo response, making interpretation more difficult. In addition, only one dose was tested. Anecdotally, the authors have found that adding oral magnesium sulphate to an existing analgesic regimen, at 500 mg three times daily, may be effective, especially if the patient complains of painful muscle spasms.
16.7 Management Strategies for Central Neuropathic Pain
If the results of the efficacy of analgesics and co-analgesics in neuropathic pain are reviewed, a clear picture emerges. Firstly, most drugs are less efficacious for central as compared to peripheral neuropathic pain. Secondly the NNT of all of the most commonly prescribed drugs are similar, between three and nine (Finnerup et al. 2010). Amitriptyline is the oldest and most active compound with an NNT just below three. The NNT have actually increased for drugs evaluated in more recent clinical trials, but this is probably related to more stringent trial methodology (Finnerup et al. 2010). Patients themselves are also important sources of information about drug efficacy, if not the most important source. They report that, after titrating up the dose, most of these drugs begin to produce side effects, making optimal dosing difficult, if not impossible, in many individuals. Drowsiness, inability to drive a car and ‘feeling like a zombie’ are common complaints. Patients with spinal cord pain also suggest that drugs such as antidepressants are less effective, when compared to other interventions, such as acupuncture (Heutink et al. 2011).
A review of all available comparison trials for neuropathic pain has been undertaken. This included industry-sponsored and other unpublished studies involved in the drug approval process (Watson et al. 2010). For instance, they reported a clinical trial with reasonable methodology (Jadad score of 3 18) which compared amitriptyline to pregabalin. For patients treated with amitriptyline, 46 % had 50 % or greater relief in pain. In comparison, 50 % or greater relief from pain was documented in 40 % of patients treated with pregabalin and 30 % of patients receiving a placebo. They used the same methodology to describe all trials comparing two or more drugs and came to the following conclusions:
1.
There is no evidence supporting the efficacy of the benzodiazepine lorazepam, the phenothiazine fluphenazine and the sodium channel-blocking agent mexiletine or carbamazepine for neuropathic pain (trigeminal neuralgia was excluded).
2.
There is no evidence for the superiority of gabapentinoids over tricyclic antidepressants, either regarding pain or adverse effects, although the nature of the latter differs with the two agents.
3.
There are nonsignificant trends suggesting the superiority of opioids over tricyclic antidepressants and gabapentinoids.
16.7.1 Combinations of Analgesics
Neuropathic pain is generated by a biochemical network of maladaptive neurons and glia. It is therefore unlikely that monotherapy will ever produce sufficient analgesia, and in practice, neuropathic pain is managed with combination of analgesics and co-analgesics. A well-known early combination was the Brompton cocktail, named after the Royal Brompton Hospital in London (Mount et al. 1976). It was a potent elixir of alcohol, cocaine, morphine and flavouring. Since many patients vomited on this concoction, antiemetics were added. Happily this type of polypharmacy is now obsolete, but the general idea of combining drugs with different but synergistic mechanisms of action is the accepted best practice for management of neuropathic pain, as recommended by the World Health Organization and many professional associations. If there is a synergistic effect between two drugs, then the dose of each individual drug and, therefore, dose-related side effects are reduced, ensuring a better balance between efficacy and safety. Take the example of drug A, which achieves analgesia at a maximal dose of at 150 mg twice daily, and drug B, which gives sufficient analgesic at a maximal dose of 25 mg twice daily. If the same efficacy is achieved by combining these two drugs at half doses, i.e. drug A at 75 mg twice daily and drug B at 12.5 mg twice daily, then this merely shows an additive effect. If, however, comparable or better analgesia is achieved with the two drugs combined at lower dosages—say drug A at 50 mg twice daily and drug B at 10 mg twice daily—then the drugs are synergistic (Smith and Argoff 2011). Another effective means of combining drugs is according to speed of onset and duration of action. If a fast-onset but short-acting analgesic is combined with a slower-onset but longer-acting agent, then the outcome may be a more rapid onset of pain relief and a longer duration of benefit. Combining analgesics also provides a means of transferring patients from one type of monotherapy to another.
There is an active debate as regards whether a stepwise approach should be adopted, i.e. increasing the first drug up to its maximal tolerated dose before then adding a second drug, or whether a combination should be used from the outset. Some argue that the first option is the only way to determine which drug is beneficial. Many favour the second approach because the sooner one reaches acceptable analgesia the better. They also reason that combining different drugs from different classes is in line with the complex pathophysiology of neuropathic pain (Harvey and Dickenson 2008).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


